







Gli ultimi aggiornamenti al Congresso AAO 2017
Nuove speranze per i pazienti affetti da maculopatie ereditarie, in base ai risultati presentati al 121mo Congresso dell’American Academy of Ophthalmology (AAO), che si è appena concluso a New Orleans (USA – 11-14 novembre 2017).
Amaurosi Congenita di Leber
I pazienti arruolati nello studio erano affetti da Amaurosi Congenita di Leber (LCA), una rara patologia genetica che, mentre le altre maculopatie ereditarie tendono a manifestarsi intorno ai 20-30 anni, insorge nella primissima infanzia e progredisce lentamente, portando infine ad una completa cecità.
Per questa, come per altre maculopatie ereditarie, non esiste al momento attuale nessuna terapia medica in grado di contrastarne il decorso.
Epidemiologia
La prevalenza tra i nati vivi dell’Amaurosi Congenita di Leber è stimata in 1/50.000-1/33.000.
L’Amaurosi Congenita di Leber rappresenta il 5% di tutte le distrofie retiniche e il 20% dei casi di cecità nei bambini in età scolare.
Quadro clinico
L’Amaurosi Congenita di Leber è caratterizzata da una grave riduzione dell’acuità visiva (≤ 20/400) o da cecità che esordisce nel primo anno di vita.
A seconda della causa genetica, si osservano risposte pupillari lente, movimenti oculari erratici, fotofobia, ipermetropia marcata, nistagmo, strabismo convergente e cheratocono.
Il segno oculo-digitale di Franceschetti (pressione del globo oculare con la punta dell’indice, pressione del bulbo oculare con il palmo della mano e strofinamento del bulbo oculare) è patognomico.
La LCA può essere causata da mutazioni nei geni responsabili di sindromi caratterizzate da ritardo dello sviluppo neurologico, disabilità intellettiva, comportamento oculomotorio di tipo aprassico (difficoltà a muovere gli occhi) e disfunzione renale.
Eziologia
L’Amaurosi Congenita di Leber è causata dalle mutazioni nei geni che codificano proteine specifiche della retina, tra i quali: GUCY2D (17p13.1), CEP290 (12q21.33), RPGRIP1 (14q11.2), RDH12 (14q24.1), SPATA7 (14q31.3), AIPL1 (17p13.1), RD3 (1q32.3), CRB1 (1q31-q32.1), CRX (19q13.3), IMPDH1 (7q31.3-q32), IQCB1 (3q21.1), KCNJ13 (2q37), LCA5 (6q14), NMNAT1 (1p36.22), e TULP1 (6p21.3). Queste mutazioni causano gravi deficit funzionali oppure sono correlate per lo più a distrofia della retina.
Le mutazioni nei geni CRX e IMPDH1 si associano ad un esordio precoce e ad una malattia grave.
I pazienti con mutazioni in GUCY2D presentano una degenerazione morfologica a progressione molto lenta e difetti per lo più funzionali.
Diagnosi
La diagnosi si basa sull’esame clinico che evidenzia una risposta pupillare lenta o pressoché assente nelle prime fasi della vita; alla fondoscopia si osserva una riduzione dei vasi retinici associata a segni variabili di degenerazione della retina (da pressoché irrilevante, fino ad un aspetto granulare generale). La diagnosi viene confermata dalla ERG in sedazione con esiti vicini alla soglia o inferiori.
L’analisi molecolare è fondamentale e viene eseguita usando un chip (APEX, che analizza una serie di mutazioni nei geni della LCA; la diagnosi viene raggiunta nel 50-70% dei casi) e il sequenziamento di seconda generazione (NGS) (che copre l’intera sequenza dei geni noti; si tratta del metodo preferenziale, che identifica fino al 90% dei pazienti). La conferma delle mutazioni individuate e l’analisi di segregazione nei genitori con il metodo Sanger confermano definitivamente la diagnosi.
Diagnosi differenziale
La diagnosi differenziale si pone con la retinite pigmentosa, la sindrome di Alström, la sindrome di Joubert, la malattia di Stargardt, la sindrome di Senior-Loken, la sindrome cono-renale e la ceroido-lipofuscinosi neuronale infantile. Quando non è possibile accedere ai test funzionali o agli esami morfologici ad alta risoluzione, i pazienti spesso vengono erroneamente diagnosticati affetti da cecità corticale.
Diagnosi prenatale
La diagnosi prenatale è offerta da laboratori specializzati alle coppie a rischio con mutazioni patogenetiche note.
Gli obiettivi della ricerca
Già nel 1997 la causa della LCA è stata identificata in determinate mutazioni del gene RPE65, che impediscono la sintesi di una proteina che consente il corretto assorbimento della vitamina A da parte delle cellule dell’epitelio pigmentato retinico.
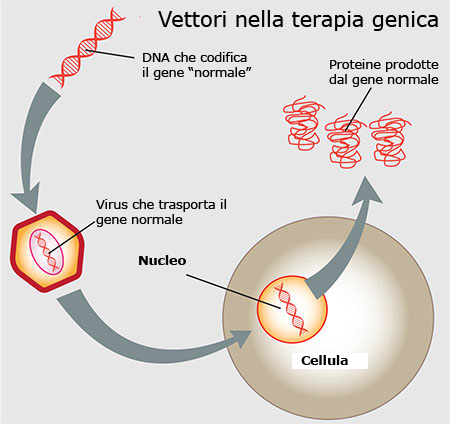
Si è, quindi, puntato a individuare il segmento di DNA che corrisponde al gene RPE65 normale e a inserirlo in un vettore costituito da un virus (AAV: adeno-associated viral vector), che viene iniettato sotto la retina dove rilascia il segmento genetico sano.
In questo modo la proteina RPE65 può essere correttamente sintetizzata e svolgere la sua attività sui recettori visivi.
I risultati
Il dr Stephen R. Russell del team di ricerca dell’Università dell’IOWA, che sta sperimentando questa terapia genetica pioneristica, riferisce che nello studio clinico di fase III 27 dei 29 pazienti trattati (il 93%) hanno ottenuto un significativo miglioramento della loro capacità visiva, tale da potersi muovere da soli in un labirinto con illuminazione medio-bassa.
È stato rilevato anche un miglioramento nella sensibilità luminosa e nella visione periferica, che costituiscono i due tipi di deficit visivo che colpiscono questi pazienti.
Chiaramente i pazienti non riacquisiscono una normale funzione visiva, ma possono distinguere le forme e percepire la luce.
Conclusioni
Non è possibile affermare con sicurezza che i risultati raggiunti persisteranno nel tempo, ma nei pazienti trattati si sono mantenuti per circa due anni. La terapia genica proposta è attualmente al vaglio della Food and And Drug Administration (FDA) e ad ottobre l’advisory committee si è unanimemente pronunciato a favore del trattamento, che potrebbe quindi ricevere l’approvazione ad inizio del 2018.
Prima terapia per Amaurosi Congenita di Leber
Sarebbe la prima terapia genica autorizzata per il trattamento di una maculopatia ereditaria e potrebbe aprire la strada a terapie analoghe per altre maculopatie dovute a mutazioni genetiche, quali la retinite pigmentosa o la maculopatia di Stargardt.
Per approfondimenti consulta la pagina The Research Road: Gene Therapy for Leber Congenital Amaurosis del National Eye Institute (NEI)
- Huang CH, Yang CM, Yang CH, Hou YC, Chen TC. Leber’s Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basel). 2021 Aug 19;12(8):1261. doi: 10.3390/genes12081261. PMID: 34440435; PMCID: PMC8392113.
- Tsang SH, Sharma T. Leber Congenital Amaurosis. Adv Exp Med Biol. 2018;1085:131-137. doi: 10.1007/978-3-319-95046-4_26. PMID: 30578499.