







Di difficile diagnosi, ma sensibile al trattamento precoce
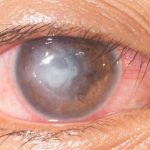
Occhi che si infiammano spesso, in pazienti di giovane età, con molti segni clinici della cheratite, ma con una particolarità: l’infiammazione della superficie oculare è associata a una riduzione significativa della capacità uditiva. L’ipotesi diagnostica da prendere in considerazione è: Sindrome di Cogan, una patologia rara ad eziologia autoimmune, così denominata perché fu identificata per la prima volta nel 1945 da David G. Cogan che descrisse i casi di 4 pazienti come “cheratite interstiziale non sifilitica accompagnata da sintomi vestibulo-uditivi”.
Insorgenza e sintomi
La malattia colpisce in prevalenza i giovani adulti, con un’età d’esordio compresa tra 20 e 30 anni, mentre è poco frequente nei bambini. L’esordio clinico è caratterizzato a livello sistemico da febbre, affaticamento e perdita di peso. Sono frequenti sintomi uditivi, qu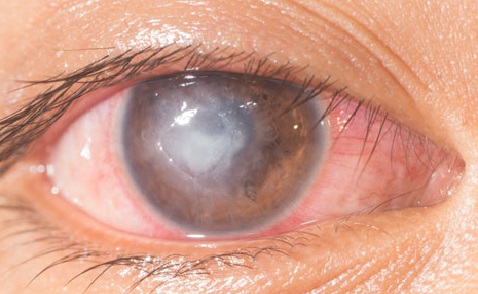 ali acufeni e riduzione dell’udito, e sintomi oculari, quali fotofobia, oscillopsia e un’infiammazione ricorrente della cornea.
ali acufeni e riduzione dell’udito, e sintomi oculari, quali fotofobia, oscillopsia e un’infiammazione ricorrente della cornea.
La cheratite interstiziale si manifesta spesso con un infiltrato a forma ad anello, che può quindi risparmiare la zona pupillare e proprio per questa ragione il primo sintomo della sindrome di Cogan può non essere un disturbo della vista.
All’esordio, la perdita dell’udito si può manifestare prima in un orecchio e poi nell’altro, e in seguito diventare bilaterale, può essere associata a disturbi dell’equilibrio, che spesso portano a formulare la diagnosi di Malattia di Ménière.
Le manifestazioni oculari, uditive e/o dell’equilibrio, inclusi i sintomi oculari, nelle fasi iniziali della malattia possono non essere concomitanti ed è proprio questo che rende molto difficile la diagnosi, se lo specialista non conosce bene il quadro clinico della Sindrome di Cogan.
Classificazione
Nel 1981 Haynes e coll. hanno proposto anche di distinguere due forme:
Sindrome di Cogan “tipica”
Questa forma è caratterizzata da ipoacusia neurosensoriale associata a cheratite interstiziale.
Sindrome di Cogan “atipica”
Questa forma è caratterizzata da ipoacusia neurosensoriale associata a infiammazione di qualsiasi parte del bulbo oculare (sclerite, episclerite, uveite). Nella forma “atipica” la cheratite interstiziale può essere presente, ma non necessariamente.
Eziologia
L’eziologia è autoimmune e l’infiammazione nell’occhio e nell’orecchio è dovuta al sistema immunitario del paziente, che produce autoanticorpi che attaccano l’orecchio interno e il tessuto oculare.
La diagnosi
Non esistono test di laboratorio specifici per diagnosticare la sindrome di Cogan. Può essere utile la titolazione degli anticorpi anti-coclea, che ha una sensibilità del 50%, per cui la negatività del test non permette di escludere la malattia, mentre la sua positività può essere di supporto alla diagnosi.
Studi recenti suggeriscono di effettuare una RMN per escludere il neurinoma del nervo acustico. Tuttavia, la RMN può non fornire risultati di rilievo.
La diagnosi è essenzialmente clinica, per cui l’anamnesi svolge un ruolo cruciale e un accurato ed approfondito esame con lampada a fessura può essere determinante.
La terapia
I corticosteroidi rappresentano il “gold standard” per la cura del trattamento della Sindrome di Cogan. I glucocorticoidi topici, associati ai cicloplegici, possono essere utilizzati per il trattamento della patologia oculare lieve e isolata mentre, quando il coinvolgimento oculare è più grave, l’udito è compromesso e sono presenti sintomi sistemici, si raccomanda l’uso dei corticosteroidi per via sistemica. Di solito la terapia sistemica a base di corticosteroidi ad alto dosaggio (somministrazione quotidiana di 1-1,5 mg/kg di prednisone) previene la sordità e mostra benefici nell’arco di 2-3 settimane. Tuttavia, i benefici a breve termine della terapia steroidea sono associati al rischio di gravi effetti collaterali. Di conseguenza, nei pazienti con malattia grave e refrattaria si raccomanda di considerare un trattamento di seconda linea con gli immunosoppressori. Gli immunosoppressori convenzionali, come metotressato, ciclofosfamide, azatiotropina o ciclosporina A, hanno scarsa efficacia, mentre cresce il numero dei casi documentati di risposta positiva all’infliximab, un inibitore del fattore di necrosi tumorale alfa (TNFalpha). I pazienti trattati con infliximab hanno mostrato un miglioramento dei sintomi cocleo-vestibolari che ha permesso una riduzione graduale del dosaggio dei corticosteroidi, con una differenza significativa rispetto ai pazienti trattati solo con steroidi o DMARD. L’uso precoce di infliximab, come trattamento di prima linea nei casi più gravi, sembra avere un’efficacia ancora maggiore. Gli impianti cocleari rappresentano un’opzione chirurgica utile nei pazienti con grave sordità neurosensoriale, che non rispondono al trattamento intensivo con gli immunosoppressori.
La prognosi dipende prevalentemente dal rischio di sordità permanente e di complicanze cardiovascolari, in particolare di insufficienza aortica. Il coinvolgimento grave degli organi interni e i decessi correlati alle complicanze cardiovascolari sono, tuttavia, rari.
Conclusione
La Sindrome di Cogan è una malattia curabile, che risponde bene al trattamento immunosoppressivo precoce. Dovrebbe, quindi, essere conosciuta molto bene dall’oculista, dall’otorinolaringoiatra, dall’internista e dal pediatra. Purtroppo, però, tutt’oggi la Sindrome di Cogan rimane una malattia rara spesso misconosciuta, perché solo pochi oculisti e otorinolaringoiatri correlano infiammazione oculare e sordità neuro- sensoriale.
Gluth Mb, Baratz Kh, Matteson El, Driscoll Cl, Cogan Syndrome: A Retrospective Review of 60 Patients Throughout a Half Century, su Mayo Clinic proceedings, 2006 Apr.
Haynes Bf, Kaiser-Kupfer Mi, Mason P, Fauci As, Cogan Syndrome: Studies in Thirteen Patients, Long-Term Follow-Up, and a Review of the Literature, su Medicine, 1980 Nov. URL
Iliescu DA, Timaru CM, Batras M, De Simone A, Stefan C. COGAN’S SYNDROME. Rom J Ophthalmol. 2015 Jan-Mar;59(1):6-13. PMID: 27373108; PMCID: PMC5729811.