







Latest updates at the 2017 AAO Congress
New hope for patients with inherited maculopathies, according to findings presented at the 121st Congress of the American Academy of Ophthalmology (AAO), which has just concluded in New Orleans (USA - 11-14 November 2017).
Leber Congenital Amaurosis
The patients enrolled in the study were suffering from Leber Congenital Amaurosis (LCA), a rare genetic disorder that, while other hereditary maculopathies tend to manifest around the age of 20-30 years, arises in very early childhood and progresses slowly, eventually leading to complete blindness.
For this, as for other hereditary maculopathies, there is currently no medical therapy capable of counteracting its course.
Epidemiology
The prevalence among live births of Leber Congenital Amaurosis is estimated at 1/50,000-1/33,000.
Leber Congenital Amaurosis accounts for 5% of all retinal dystrophies and 20% of cases of blindness in school-age children.
Clinical picture
Leber's congenital amaurosis is characterised by a severe reduction in visual acuity (≤ 20/400) or blindness that begins in the first year of life.
Depending on the genetic cause, slow pupillary responses, erratic eye movements, photophobia, marked hypermetropia, nystagmus, convergent strabismus and keratoconus are observed.
Franceschetti's oculo-digital sign (pressing the eyeball with the tip of the index finger, pressing the eyeball with the palm of the hand and rubbing the eyeball) is pathognomic.
LCA can be caused by mutations in the genes responsible for syndromes characterised by neurodevelopmental delay, intellectual disability, apraxic-like oculomotor behaviour (difficulty moving the eyes) and renal dysfunction.
Aetiology
Leber Congenital Amaurosis is caused by mutations in genes encoding specific retinal proteins, including: GUCY2D (17p13.1), CEP290 (12q21.33), RPGRIP1 (14q11.2), RDH12 (14q24.1), SPATA7 (14q31.3), AIPL1 (17p13.1), RD3 (1q32.3), CRB1 (1q31-q32.1), CRX (19q13.3), IMPDH1 (7q31.3-q32), IQCB1 (3q21.1), KCNJ13 (2q37), LCA5 (6q14), NMNAT1 (1p36.22), and TULP1 (6p21.3). These mutations cause severe functional deficits or are mostly related to retinal dystrophy.
Mutations in genes CRX e IMPDH1 are associated with early onset and severe disease.
Patients with mutations in GUCY2D have very slowly progressing morphological degeneration and mostly functional defects.
Diagnosis
The diagnosis is based on the clinical examination, which shows a slow or almost absent pupillary response in the early stages of life; at fundoscopy, a reduction of the retinal vessels is observed associated with variable signs of retinal degeneration (from almost negligible to a general granular appearance). The diagnosis is confirmed by ERG under sedation with outcomes close to threshold or below.
Molecular analysis is crucial and is performed using a chip (APEX, which analyses a number of mutations in the LCA genes; the diagnosis is reached in 50-70% of cases) and second-generation sequencing (NGS) (which covers the entire sequence of known genes; this is the preferred method, which identifies up to 90% of patients). Confirmation of the identified mutations and segregation analysis in the parents by the Sanger method definitively confirm the diagnosis.
Differential diagnosis
The differential diagnosis arises with retinitis pigmentosa, Alström's syndrome, Joubert's syndrome, Stargardt's disease, Senior-Loken syndrome, cono-renal syndrome and infantile neuronal ceroid-lipofuscinosis. When functional tests or high-resolution morphological examinations are not available, patients are often misdiagnosed as having cortical blindness.
Prenatal diagnosis
Prenatal diagnosis is offered by specialised laboratories to at-risk couples with known pathogenic mutations.
Research objectives
As early as 1997, the cause of LCA was identified as certain mutations in the RPE65 genewhich prevent the synthesis of a protein that enables the proper absorption of vitamin A by the cells of the retinal pigment epithelium.
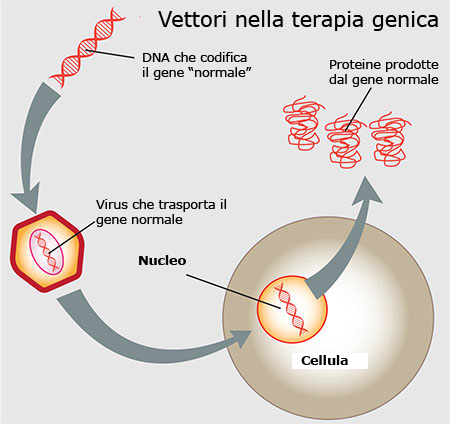
The aim was therefore to identify the DNA segment corresponding to the normal RPE65 gene and insert it into a vector consisting of a virus (AAV: adeno-associated viral vector), which is injected under the retina where it releases the healthy genetic segment.
In this way, the RPE65 protein can be correctly synthesised and perform its activity on visual receptors.
The results
Dr. Stephen R. Russell of the IOWA University research team, which is testing this pioneering gene therapy, reports that in the phase III clinical trial, 27 of the 29 patients treated (the 93%) achieved a significant improvement in their visual acuity, such that they were able to move around a maze with low to medium illumination on their own.
An improvement in light sensitivity and peripheral vision, the two types of visual impairment affecting these patients, was also noted.
Clearly, patients do not regain normal visual function, but they can distinguish shapes and perceive light.
Conclusions
It is not possible to say with certainty that the results achieved will persist over time, but in treated patients they have been maintained for about two years. The proposed gene therapy is currently being evaluated by the Food and And Drug Administration (FDA) and in October theadvisory committee was unanimously in favour of the treatment, which could therefore receive approval in early 2018.
First therapy for Leber Congenital Amaurosis
It would be the first gene therapy authorised for the treatment of a hereditary maculopathy and could pave the way for similar therapies for other maculopathies due to genetic mutations, such as retinitis pigmentosa or Stargardt's maculopathy.
For more information see The Research Road: Gene Therapy for Leber Congenital Amaurosis of the National Eye Institute (NEI)
- Huang CH, Yang CM, Yang CH, Hou YC, Chen TC. Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basel). 2021 Aug 19;12(8):1261. doi: 10.3390/genes12081261. PMID: 34440435; PMCID: PMC8392113.
- Tsang SH, Sharma T. Leber Congenital Amaurosis. Adv Exp Med Biol. 2018;1085:131-137. doi: 10.1007/978-3-319-95046-4_26. PMID: 30578499.