







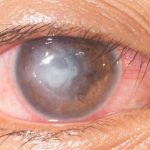
Difícil de diagnosticar, pero sensible al tratamiento precoz
Ojos que a menudo se inflaman, en pacientes jóvenes, con muchos de los signos clínicos de la queratitis, pero con una peculiaridad: la inflamación de la superficie ocular se asocia a una reducción significativa de la capacidad auditiva. La hipótesis diagnóstica a considerar es: el síndrome de Cogan, una enfermedad rara de etiología autoinmune, llamada así porque fue identificada por primera vez en 1945 por David G. Cogan que describió los casos de 4 pacientes como "Queratitis intersticial no sifilítica acompañada de síntomas vestíbulo-auditivos"..
Inicio y síntomas
La enfermedad afecta predominantemente a adultos jóvenes, con una edad de aparición entre los 20 y los 30 años, mientras que es infrecuente en niños. El inicio clínico se caracteriza sistémicamente por fiebre, fatiga y pérdida de peso. Son frecuentes síntomas auditivos, qu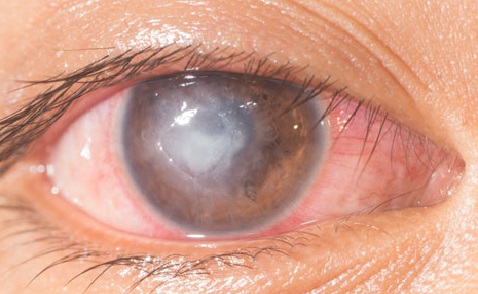 acúfenos y disminución de la audición, y síntomas ocularescomo fotofobia, oscilopsia e inflamación recurrente de la córnea.
acúfenos y disminución de la audición, y síntomas ocularescomo fotofobia, oscilopsia e inflamación recurrente de la córnea.
La queratitis intersticial suele manifestarse con un infiltrado en forma de anillo, que por lo tanto puede prescindir de la zona pupilar y, por esta misma razón, el primer síntoma del síndrome de Cogan puede no ser una alteración visual.
Al principio, la pérdida de audición puede producirse primero en un oído y luego en el otro, y más tarde hacerse bilateral. Puede ir asociada a trastornos del equilibrio, que a menudo conducen al diagnóstico de la enfermedad de Ménière.
Las manifestaciones oculares, auditivas y/o del equilibrio, incluidos los síntomas oculares, en las primeras fases de la enfermedad pueden no ser concomitantes y es precisamente esto lo que hace que el diagnóstico sea muy difícil si el especialista no está familiarizado con el cuadro clínico del síndrome de Cogan.
Clasificación
En 1981, Haynes et al. también propusieron distinguir dos formas:
Típico síndrome de Cogan
Esta forma se caracteriza por una pérdida de audición neurosensorial asociada a una queratitis intersticial.
Síndrome de Cogan "atípico
Esta forma se caracteriza por una pérdida de audición neurosensorial asociada a una inflamación de cualquier parte del globo ocular (escleritis, epiescleritis, uveítis). En la forma "atípica", puede haber queratitis intersticial, pero no necesariamente.
Etiología
La etiología es autoinmune y la inflamación en el ojo y el oído se debe al sistema inmunitario del paciente, que produce autoanticuerpos que atacan el oído interno y el tejido ocular.
Diagnóstico
No existen pruebas de laboratorio específicas para diagnosticar el síndrome de Cogan. La valoración de anticuerpos anti-Cogan, que tiene una sensibilidad de 50%, puede ser útil, de modo que la negatividad de la prueba no permite descartar la enfermedad, mientras que su positividad puede apoyar el diagnóstico.
Estudios recientes sugieren la RM para descartar un neurinoma del nervio auditivo. Sin embargo, la IRM puede no ofrecer resultados relevantes.
El diagnóstico es esencialmente clínico, por lo que la historia clínica desempeña un papel crucial y un examen minucioso y exhaustivo con lámpara de hendidura puede ser decisivo.
Terapia
Los corticosteroides representan el "patrón oro" para el tratamiento del síndrome de Cogan. Los glucocorticoides tópicos, combinados con ciclopléjicos, pueden utilizarse para el tratamiento de la patología ocular leve y aislada, mientras que, cuando la afectación ocular es más grave, la audición está alterada y hay síntomas sistémicos, se recomiendan los corticoesteroides sistémicos. Por lo general, el tratamiento sistémico con dosis altas de corticosteroides (administración diaria de 1-1,5 mg/kg de prednisona) previene la sordera y muestra beneficios en 2-3 semanas. Sin embargo, los beneficios a corto plazo del tratamiento con corticoides se asocian al riesgo de efectos secundarios graves. Por consiguiente, en pacientes con enfermedad grave y refractaria, se recomienda considerar el tratamiento de segunda línea con inmunosupresores. Los inmunosupresores convencionales, como el metotrexato, la ciclofosfamida, la azatiotropina o la ciclosporina A, tienen una eficacia escasa, mientras que está aumentando el número de casos documentados de respuesta positiva al infliximab, un inhibidor del factor de necrosis tumoral alfa (TNFalfa). Los pacientes tratados con infliximab mostraron una mejoría de los síntomas cócleo-vestibulares que permitió una reducción gradual de la dosis de corticosteroides, con una diferencia significativa en comparación con los pacientes tratados únicamente con esteroides o DMARD. El uso precoz de infliximab, como tratamiento de primera línea en los casos más graves, parece tener una eficacia aún mayor. Los implantes cocleares representan una opción quirúrgica útil en pacientes con sordera neurosensorial grave que no responden al tratamiento intensivo con inmunosupresores.
El pronóstico depende principalmente del riesgo de sordera permanente y de complicaciones cardiovasculares, en particular insuficiencia aórtica. Sin embargo, la afectación grave de órganos internos y las muertes relacionadas con complicaciones cardiovasculares son poco frecuentes.
Conclusión
El síndrome de Cogan es una enfermedad curableque responde bien a un tratamiento inmunosupresor precoz. Por lo tanto, debería ser muy bien conocida por el oftalmólogo, el otorrinolaringólogo, el internista y el pediatra. Sin embargo, por desgracia, a día de hoy el síndrome de Cogan sigue siendo una enfermedad rara que a menudo se malinterpreta, porque sólo unos pocos oftalmólogos y otorrinolaringólogos correlacionan la inflamación ocular y la sordera neurosensorial.
Gluth Mb, Baratz Kh, Matteson El, Driscoll Cl, Síndrome de Cogan: revisión retrospectiva de 60 pacientes a lo largo de medio sigloen Mayo Clinic proceedings, 2006 abr.
Haynes Bf, Kaiser-Kupfer Mi, Mason P, Fauci As, Síndrome de Cogan: estudios en trece pacientes, seguimiento a largo plazo y revisión de la literatura, en Medicina, 1980 Nov. URL
Iliescu DA, Timaru CM, Batras M, De Simone A, Stefan C. SÍNDROME DE COGAN. Rom J Ophthalmol. 2015 Jan-Mar;59(1):6-13. PMID: 27373108; PMCID: PMC5729811.