







Las últimas actualizaciones en el Congreso de la AAO 2017
Nuevas esperanzas para los pacientes con maculopatías hereditarias, según los hallazgos presentados en el 121º Congreso de la Academia Americana de Oftalmología (AAO), que acaba de concluir en Nueva Orleans (Estados Unidos - 11-14 de noviembre de 2017).
Amaurosis Congénita de Leber
Los pacientes incluidos en el estudio padecían Amaurosis Congénita de Leber (ACL), un trastorno genético poco frecuente que, mientras que otras maculopatías hereditarias tienden a manifestarse en torno a los 20-30 años de edad, surge en la primera infancia y progresa lentamente, hasta desembocar en la ceguera total.
Para ésta, como para otras maculopatías hereditarias, no existe actualmente ninguna terapia médica capaz de contrarrestar su curso.
Epidemiología
La prevalencia entre los nacidos vivos de la Amaurosis Congénita de Leber se estima en 1/50.000-1/33.000.
La Amaurosis Congénita de Leber representa el 5% de todas las distrofias de retina y el 20% de los casos de ceguera en niños en edad escolar.
Cuadro clínico
La amaurosis congénita de Leber se caracteriza por una reducción grave de la agudeza visual (≤ 20/400) o ceguera que comienza en el primer año de vida.
Dependiendo de la causa genética, se observan respuestas pupilares lentas, movimientos oculares erráticos, fotofobia, hipermetropía marcada, nistagmo, estrabismo convergente y queratocono.
El signo óculo-digital de Franceschetti (presionar el globo ocular con la punta del dedo índice, presionar el globo ocular con la palma de la mano y frotar el globo ocular) es patognomónico.
La ECV puede estar causada por mutaciones en los genes responsables de síndromes caracterizados por retraso del neurodesarrollo, discapacidad intelectual, comportamiento oculomotor apráxico (dificultad para mover los ojos) y disfunción renal.
Etiología
La Amaurosis Congénita de Leber está causada por mutaciones en genes que codifican proteínas retinianas específicas, entre las que se incluyen: GUCY2D (17p13.1), CEP290 (12q21.33), RPGRIP1 (14q11.2), RDH12 (14q24.1), SPATA7 (14q31.3), AIPL1 (17p13.1), RD3 (1q32.3), CRB1 (1q31-q32.1), CRX (19q13.3), IMPDH1 (7q31.3-q32), IQCB1 (3q21.1), KCNJ13 (2q37), LCA5 (6q14), NMNAT1 (1p36.22), y TULP1 (6p21.3). Estas mutaciones causan graves déficits funcionales o están relacionadas sobre todo con la distrofia retiniana.
Mutaciones en los genes CRX e IMPDH1 se asocian con una aparición precoz y una enfermedad grave.
Los pacientes con mutaciones en GUCY2D presentan una degeneración morfológica de progresión muy lenta y, en su mayoría, defectos funcionales.
Diagnóstico
El diagnóstico se basa en el examen clínico, que muestra una respuesta pupilar lenta o casi ausente en las primeras etapas de la vida; en la fundoscopia, se observa una reducción de los vasos retinianos asociada a signos variables de degeneración de la retina (desde casi inapreciable hasta un aspecto granular general). El diagnóstico se confirma mediante ERG bajo sedación con resultados próximos al umbral o inferiores.
El análisis molecular es crucial y se realiza mediante un chip (APEX, que analiza una serie de mutaciones en los genes del ACV; el diagnóstico se alcanza en el 50-70% de los casos) y secuenciación de segunda generación (NGS) (que abarca toda la secuencia de genes conocidos; es el método preferido, que identifica hasta el 90% de los pacientes). La confirmación de las mutaciones identificadas y el análisis de segregación en los padres mediante el método Sanger confirman definitivamente el diagnóstico.
Diagnóstico diferencial
El diagnóstico diferencial se plantea con la retinosis pigmentaria, el síndrome de Alström, el síndrome de Joubert, la enfermedad de Stargardt, el síndrome de Senior-Loken, el síndrome cono-renal y la ceroidolipofuscinosis neuronal infantil. Cuando no se dispone de pruebas funcionales o exámenes morfológicos de alta resolución, los pacientes suelen ser diagnosticados erróneamente de ceguera cortical.
Diagnóstico prenatal
El diagnóstico prenatal lo ofrecen laboratorios especializados a las parejas de riesgo con mutaciones patogénicas conocidas.
Objetivos de la investigación
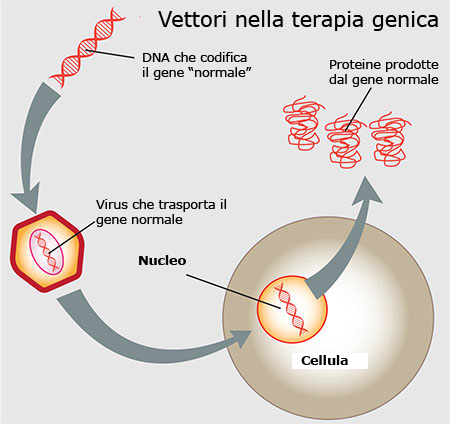
Ya en 1997, se identificó que la causa de la ECV eran ciertas mutaciones en el Gen RPE65que impiden la síntesis de una proteína que permite la correcta absorción de la vitamina A por las células del epitelio pigmentario de la retina.
El objetivo era, por tanto, identificar el segmento de ADN correspondiente al gen RPE65 normal e insertarlo en un vector consistente en un virus (AAV: vector viral adenoasociado), que se inyecta bajo la retina, donde libera el segmento genético sano.
De este modo, la proteína RPE65 puede sintetizarse correctamente y realizar su actividad en los receptores visuales.
Los resultados
El Dr. Stephen R. Russell, del equipo de investigación de la Universidad de IOWA, que está probando esta terapia génica pionera, informa de que en el ensayo clínico de fase III 27 de los 29 pacientes tratados (los 93%) lograron una mejora significativa de su agudeza visual, hasta el punto de poder desplazarse solos por un laberinto con iluminación baja o media.
También se observó una mejora de la sensibilidad a la luz y de la visión periférica, los dos tipos de deficiencias visuales que afectan a estos pacientes.
Evidentemente, los pacientes no recuperan la función visual normal, pero pueden distinguir formas y percibir la luz.
Conclusiones
No es posible afirmar con certeza que los resultados obtenidos persistan en el tiempo, pero en los pacientes tratados se han mantenido durante unos dos años. La terapia génica propuesta está siendo evaluada actualmente por la Food and Drug Administration (FDA) y en octubre lacomité consultivo se pronunció unánimemente a favor del tratamiento, que podría así recibir la aprobación a principios de 2018.
Primera terapia para la amaurosis congénita de Leber
Sería la primera terapia génica autorizada para el tratamiento de una maculopatía hereditaria y podría allanar el camino a terapias similares para otras maculopatías debidas a mutaciones genéticas, como la retinosis pigmentaria o la maculopatía de Stargardt.
Para más información El camino de la investigación: terapia génica para la amaurosis congénita de Leber del Instituto Nacional del Ojo (NEI)
- Huang CH, Yang CM, Yang CH, Hou YC, Chen TC. Leber's Congenital Amaurosis: Current Concepts of Genotype-Phenotype Correlations. Genes (Basilea). 2021 Aug 19;12(8):1261. doi: 10.3390/genes12081261. PMID: 34440435; PMCID: PMC8392113.
- Tsang SH, Sharma T. Amaurosis congénita de Leber. Adv Exp Med Biol. 2018;1085:131-137. doi: 10.1007/978-3-319-95046-4_26. PMID: 30578499.