







La degeneración macular asociada a la edad (DMAE) es la principal causa de reducción grave de la agudeza visual en personas mayores de 60 años en el mundo occidental. Es una enfermedad compleja que reconoce varios factores de riesgo, como la edad, el sexo, la raza, la exposición a la luz, la dieta, el tabaquismo y las enfermedades vasculares.
Recientemente, otros elementos parecen desempeñar un papel importante en la patogénesis de la enfermedad: variaciones en las secuencias de ADN que codifican los factores del complemento, modificaciones de la TMP-3 y activación de citoquinas inflamatorias. El conocimiento de nuevos factores implicados en la patogénesis de esta enfermedad puede allanar en el futuro el camino hacia nuevas estrategias terapéuticas para combatir la DMAE.
INTRODUCCIÓN
La degeneración macular asociada a la edad (DMAE) es una enfermedad degenerativa que afecta a la región macular en personas mayores de 55 años. Su gravedad y frecuencia la convierten en la principal causa de reducción grave de la agudeza visual, denominada "ceguera legal", en personas mayores de 60 años en el mundo occidental. La DMAE puede dividirse clínicamente en una forma seca (seca), la más frecuente y menos grave, y una forma exudativa (húmeda). Esta última representa aproximadamente 20% de la DMAE, pero es la causa de aproximadamente 90% de los casos graves de reducción irreversible de la agudeza visual que siempre se relacionan con esta enfermedad. Esta enfermedad conlleva una pérdida progresiva de la porción central del campo visual, con preservación de la periférica, lo que supone dejar de poder leer, conducir, reconocer caras de personas, números de autobús, etc.
Según estimaciones bastante recientes, cada año aparecen unos 500.000 nuevos casos de DMAE exudativa en todo el mundo y esta cifra, con el aumento de la esperanza media de vida, tenderá a aumentar drásticamente en las próximas décadas. Se calcula que actualmente padecen la enfermedad 8,5 millones de estadounidenses, y para 2020 se esperan unos 7,5 millones de nuevos casos en Estados Unidos.
La degeneración macular asociada a la edad es una enfermedad compleja de estudiar debido a la posible implicación de diversos factores, como los demográficos, ambientales, los riesgos asociados a la edad, el sexo, la raza, la exposición a la luz, la dieta, el tabaquismo y las enfermedades cardiovasculares.
La degeneración macular y/o las distrofias maculares pueden clasificarse en formas juveniles y seniles. En cuanto a la genética molecular de las distrofias maculares juveniles, la investigación de los últimos 10 años ha contribuido significativamente a la comprensión de estas formas. Se han identificado las características clínicas y los genes implicados. Gran parte del progreso en la comprensión de las distrofias maculares juveniles está relacionado con la aparición temprana de la enfermedad. En conjunto, estos estudios han permitido identificar los numerosos genes implicados en el proceso visual.
Recientemente se ha descubierto que los polimorfismos de un solo nucleótido y las variaciones en la secuencia del ADN, que se encuentran dentro del gen del factor H del complemento (CFH), están fuertemente asociados con el desarrollo de la DMAE en caucásicos. Los mismos autores descubrieron la implicación de otros genes en la DMAE, entre ellos el gen VEGF y dos genes relacionados con el metabolismo de los lípidos. Un polimorfismo de un solo nucleótido del CFH, Tyr402His, se asoció con aproximadamente 50% de los casos de DMAE. Otro gen implicado en la cascada del complemento se ha asociado con la patogénesis de la DMAE. Este gen, localizado en el cromosoma 6, produce una proteína llamada Factor B, que activa el complemento. Un tercer gen implicado (LOC387715) se ha mapeado en el cromosoma 10; el riesgo de desarrollar DMAE parece estar relacionado con la variación de un solo aminoácido en la proteína codificada, que no está relacionada con la cascada del complemento. Los análisis genéticos indican que el efecto del LOC387715 es independiente del del factor H, pero igualmente importante, contribuyendo posiblemente a casi 40% de los casos de DMAE. Pericak-Vance et al. mostraron una relación entre la variante de alto riesgo de LOC387715 y el tabaquismo, un importante factor de riesgo ambiental de la DMAE. Los datos comunicados hasta ahora muestran lo importante que es que la investigación científica pueda, en términos de prevención, desarrollar una nueva terapia capaz de detener el dramático desarrollo de esta enfermedad verdaderamente discapacitante.
Por lo que respecta a la AWD en seco, existen varios estudios:
- Estudio sobre las enfermedades oculares relacionadas con la edad (AREDS):
Ensayo multicéntrico estadounidense creado para evaluar si las vitaminas, los antioxidantes y/o los minerales reducen el desarrollo y/o la progresión de la enfermedad.
- Ensayo clínico sobre el tratamiento profiláctico de la degeneración macular asociada a la edad (PTAMPD):
Estudio piloto aleatorizado sobre la eficacia del tratamiento con láser de las drusas blandas en el polo posterior, que demostró, a los dos años, una reducción de las drusas en el 70% de los sujetos tratados y, en consecuencia, una mejora de la agudeza visual. En el caso de la DMAE exudativa, al tratamiento tradicional con láser basado en la destrucción de las zonas neovasculares se han añadido nuevas propuestas que provocan daños anatómicos y funcionales en la retina tratada:
- Tratamiento focal de los vasos alimentadores: basado en la fotocoagulación de baja intensidad con láser argón y/o láser diodo (infrarrojo) de los neovasos aferentes (vasos alimentadores) de las membranas neovasculares, identificados mediante angiografía dinámica con fluoresceína sódica y verde de indocianina con oftalmoscopios de barrido láser.
- Terapia térmica transpupilar (TTT): está indicada principalmente para las formas "ocultas", aprovecha el efecto térmico del láser de diodo para provocar la oclusión de las membranas neovasculares sin destruir la retina suprayacente. No obstante, se trata de una técnica que todavía necesita más estudios y modificaciones; la dificultad de esta técnica está relacionada con la falta de datos sobre la dosis correcta que se debe utilizar. Se ha introducido para el tratamiento de pequeños melanomas de la coroides del polo posterior.
- Extirpación quirúrgica: Los resultados son bastante decepcionantes en la DMAE debido al daño iatrogénico en el epitelio pigmentario de la retina. Se obtienen mejores resultados con la "rotación" retiniana (Machemer, Erkhardt y Todt) y la "translocación limitada" (De Juan, Tano), que desplaza la región foveal de la membrana subyacente para poder fotocoagular esta última.
- Radioterapia: tiene por objeto impedir la proliferación de células endoteliales en las neovasos coroideas e inducir su cierre mediante radiaciones ionizantes. Sin embargo, los ensayos clínicos aleatorizados (DAR) no han dado buenos resultados.
- Terapia fotodinámica (TFD): Antes de su aparición, había pocas opciones de intervención para las neovascularizaciones coroideas subfoveales. Tres ensayos multicéntricos, TAP, VIP y VIT (este último realizado en Italia y coordinado por la Clínica Oftalmológica del Hospital San Paolo de Milán) han demostrado su eficacia.
- Terapia farmacológica: fármacos con cortisona (acetónido de triamcinolona) y fármacos con acción antiangiogénica y angiostática como los anti-VEGF AVASTIN, MACUGEN y LUCENTIS, todos ellos administrados por vía intravítrea.
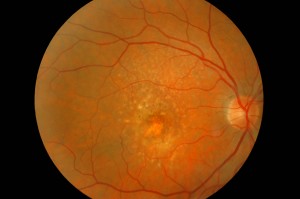
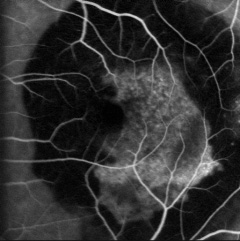
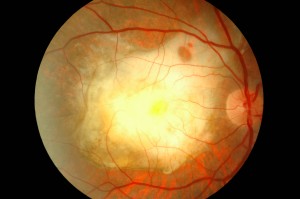
CARACTERÍSTICAS CLÍNICAS
La DMAE, como ya se ha mencionado, se ha clasificado en dos condiciones clínicas: una forma seca y una forma húmeda. La neovascularización coroidea es característica de la forma húmeda, un estadio que se encuentra en aproximadamente 10% de los casos. Aunque ambas formas de DMAE pueden causar pérdida visual, la forma húmeda está implicada en aproximadamente 90% de los casos de pérdida visual grave. También se acepta generalmente que la forma húmeda de la DMAE precede y se desarrolla a partir de la forma seca. En cambio, la forma seca es mucho más frecuente y se caracteriza clínicamente por la presencia de drusas maculares, que son depósitos localizados entre el EPR y la membrana de Bruch, y por la atrofia geográfica, caracterizada por la muerte celular del EPR con atrofia de los fotorreceptores suprayacentes.
La DMAE no suele ser clínicamente evidente antes de los 50 años. Según los resultados de las autopsias, la DMAE se encuentra en 33% de los ojos de las personas mayores de 65 años y en aproximadamente 30% de las personas mayores de 75 años. La prevalencia de la enfermedad oscila entre 1,6% en individuos de 52 a 64 años y 27,9% en mayores de 75 años. La prevalencia de la DMAE es insignificante a los 50 años y alcanza los 6% a los 80 años.
Estas discrepancias pueden explicarse, en parte, por el hecho de que el ojo, al igual que otros órganos, experimenta un proceso de envejecimiento normal independiente de la DMAE, lo que, a diferencia de las distrofias maculares juveniles, puede dificultar el diagnóstico. Además, la DMAE está asociada a enfermedades cardiovasculares y otros trastornos importantes, por lo que es posible que muchos individuos potencialmente afectados mueran de enfermedades cardiovasculares antes de que se manifieste la DMAE, lo que reduciría la prevalencia de la enfermedad en personas de 80 años.
FACTORES DE RIESGO DEMOGRÁFICOS Y AMBIENTALES DE LA DMLE
La edad, el sexo, la raza, la exposición a la luz, las enfermedades cardiovasculares concomitantes, la dieta y el tabaquismo son posibles factores de riesgo de la DMAE. La prevalencia de la DMAE es mayor en la población blanca que en la no blanca. Existen estudios contradictorios sobre el efecto de la exposición crónica a la luz (ultravioleta o visible) en la retina.
DRUSAS COMO MARCADORES INMUNOLÓGICOS DE LA DMLE
Los componentes moleculares y celulares de las drusas se han analizado exhaustivamente. Gran parte del material presente en las drusas es sintetizado por células que normalmente se encuentran en el ojo, pero parte del material procede de fuentes extraoculares. Por ejemplo, el complemento, los lípidos y las lipoproteínas B y E son componentes comunes de las drusas oculares y las placas ateroscleróticas, lo que sugiere que los mismos procesos bioquímicos e inmunológicos pueden estar implicados en ambas enfermedades. Por ejemplo, la beta amiloide, uno de los principales componentes inflamatorios de las placas de Alzheimer, también se encuentra en las drusas. Hageman et al. propusieron que las drusas pueden ser el producto de una respuesta inflamatoria localizada tras un daño por EPR en la que intervienen antígenos HLA y el sistema del complemento. La hipótesis se basa en la observación de drusas en la GNMPII, una enfermedad renal en la que la disfunción inmunológica mediada por el complemento conduce a la insuficiencia renal. En esta enfermedad, las drusas son idénticas a las de la DMAE.
Las drusas son depósitos focales de material extracelular, situados entre la membrana basal del EPR y la capa de colágeno de la membrana de Bruch. Las drusas son importantes factores de riesgo e indicadores biológicos de la degeneración macular asociada a la edad.
Se observan con frecuencia en individuos mayores de 60 años y en el contexto clínico de la DMAE. El tamaño, número, extensión y confluencia de las drusas son determinantes importantes en el riesgo de desarrollar DMAE (Pauleikhoff et al., 1990). La presencia de drusas blandas, grandes y/o confluentes se correlaciona con la aparición de neovascularización coroidea, con un riesgo relativo de 2,1 en ojos con cinco o más drusas y de 1,5 en ojos con una o más drusas grandes (Macular Photocoagulation Study Group, 1993).
La presencia de drusas se asocia a diversos déficits visuales que se producen antes de la pérdida de agudeza visual: cambios en la sensibilidad al color y al contraste, recuperación funcional macular, sensibilidad visual central y sensibilidad al contraste espaciotemporal (Frennesson et al., 1995; Holz et al., 1995; Midena et al., 1997, 1994; Stangos et al., 1995; Tolentino et al., 1994).
Se han realizado varios estudios con el objetivo de determinar la composición de las drusas, en la creencia de que la comprensión de la constitución de un depósito relacionado con la enfermedad podría proporcionar nueva información sobre el propio proceso patogénico.
Lípidos
Donders observó por primera vez la presencia de lípidos en las drusas en 1854. Utilizando técnicas histoquímicas y enzimáticas, llegó a la conclusión de que el componente lipídico de las drusas probablemente estaba formado por moléculas de glicolípidos, cerebrósidos y/o gangliósidos (Wolter y Falls, 1962; Farkas et al., 1971; Pauleikhoff et al., 1992). Pauleikhoff et al. (1992) sugirieron la existencia de distintas clases de drusas, algunas con características más "hidrofílicas" y otras más "hidrofóbicas". Holz et al. (1994) encontraron una mayor concentración de lípidos en la región macular que en las regiones periféricas de la membrana de Bruch; las características de estos lípidos sugerirían su origen celular más que vascular. Por el contrario, Curcio et al. (2001) describen la presencia de formas esterificadas y no esterificadas de colesterol en la membrana de Bruch y en las drusas: estos autores plantean la hipótesis de que la considerable cantidad de colesterol esterificado en las drusas puede implicar un origen vascular (plasmático) de los lípidos asociados a las drusas y a la membrana de Bruch. Los estudios de Haimovici et al. (2001) demuestran la presencia de ésteres de colesterol en las drusas.
A pesar de los numerosos estudios realizados, la fuente (celular, vascular o combinada) de los lípidos asociados a las drusas sigue siendo un enigma. Sin embargo, Hageman et al. comprobaron que los genes de algunas lipoproteínas asociadas a las drusas (como la lipoproteína E) se transcriben localmente, mientras que otras moléculas (como el componente P del amiloide) parecen tener un origen hepático y llegarían a la membrana de Bruch a través de la circulación. Los lípidos asociados a las drusas también podrían tener un origen heterogéneo.
Carbohidratos
Farkas et al. admitieron por primera vez la existencia de glicolípidos dentro de las drusas (1971). Kliffen et al. (1994; 1996), Farkas et al. (1971; 1996) identificaron glicoconjugados ricos en glicosaminoglicanos en depósitos dentro de la membrana de Bruch.
Mullins et al. (1997) identificaron los glucocompuestos con terminaciones consistentes en glucosa/manosa, N-acetilglucosamina, ácido siálico como los carbohidratos más comúnmente representados en las drusas, tanto blandas como duras.
Cuando se eliminaron los residuos de ácido siálico de las terminaciones carbohidratadas asociadas a las drusas mediante neuraminidasa, se encontraron disacáridos de galactosamina (Mullins y Hageman, 1999). Estos disacáridos estaban confinados en un núcleo central formado por glicoproteínas con carbohidratos unidos mediante enlaces O-glicosídicos. La presencia de estos dominios centrales en las drusas indica que el núcleo desempeña un papel en las primeras fases del desarrollo de las drusas, similar al crecimiento de una perla en la concha de una ostra.
Proteína
Los componentes proteínicos de las drusas identificados por primera vez mediante métodos inmunohistoquímicos incluían la ubiquitina (Loeffler y Mangini, 1997), las integrinas (Brem et al., 1994), los inhibidores tisulares de las metaloproteasas (Fariss et al, 1997), productos finales de glicosilación (Ishibashi et al., 1998), beta-amiloide (Loeffler et al., 1995), fibronectina (Pauleikhoff et al., 1992) y C1q (van der Schaft et al., 1993). En los últimos años, se han identificado la vitronectina (Hageman et al., 1999), el componente P del amiloide, la apolipoproteína E, el factor X, las cadenas lambda de las inmunoglobulinas, factores de activación del complemento como el complejo C5b-9 y antígenos MHC de clase II (Johnson et al., 2000; Mullins et al., 2000; Mullins y Hageman, 1997). La heterogeneidad molecular no se corresponde con fenotipos definidos ultraestructural o clínicamente (Hageman y Mullins, 1999).
Prácticamente todas las proteínas descubiertas recientemente en las drusas están asociadas de algún modo a la inflamación y/o a otros procesos asociados al sistema inmunitario. Algunas son proteínas clásicas de la fase aguda, mientras que otras son componentes de la cascada del complemento o inhibidores de la vía de ataque de la membrana del complemento. Otras están asociadas a la activación inmunológica, la coagulación y la fibrinólisis. Además, muchas de estas moléculas son comunes en depósitos patológicos asociados a otras enfermedades, como la enfermedad de Alzheimer, la aterosclerosis, la elastosis, la amiloidosis y la glomerulonefritis (Mullins et al., 2000), por lo que se pensó que era posible que en su formación estuvieran implicadas vías patogenéticas comunes.
Componentes celulares
Según los modelos tradicionales de formación de drusas, todo el material celular que contienen es de origen EPR. De hecho, en las drusas "iniciales" pueden encontrarse fragmentos celulares y orgánulos derivados de la EPR, incluso células enteras. Además, se han descrito vesículas de EPR que se extienden a través de la membrana basal de la EPR dentro de las drusas o en los lugares de su formación posterior (Ishibashi et al., 1986). Los componentes de la EPR, como la lipofuscina y la melanina, pueden observarse a veces en drusas pequeñas y tempranas.
Una observación nueva y potencialmente muy significativa es que las moléculas normalmente asociadas a las células, incluidos los antígenos HLA-DR y CD específicos, se asocian a las drusas. Estas moléculas suelen localizarse en dominios solitarios, similares a núcleos, en el contexto de las drusas (Mullins et al., 2000). Recientes análisis inmunofenotípicos han documentado que estos núcleos derivan de extensiones celulares de células dendríticas coroideas, potentes células presentadoras de antígenos asociadas a diversos procesos de inmunomodulación. Además, estos procesos de células dendríticas están típicamente asociados a las vesículas EPR ya descritas. Estos datos sugieren por primera vez que el proceso de formación de drusas puede estar mediado por células y que vías inmunitarias específicas pueden desempeñar papeles significativos en estos acontecimientos.
ARNm
Muchas de las moléculas asociadas a las drusas, recientemente identificadas mediante métodos inmunohistoquímicos, se sintetizan principalmente en el hígado. Su salida de los capilares de la coroides y posterior agregación a lo largo de la membrana de Bruch es un posible inicio hacia su deposición dentro de las drusas. Sin embargo, ciertos tipos de células locales de la neurorretina, el EPR y/o la coroides, que se encuentran cerca de la membrana de Bruch, también pueden tener la capacidad de sintetizar parte de estas moléculas. Recientemente se han obtenido pruebas de la transcripción génica de algunas moléculas asociadas a drusas en la neurorretina, el EPR y la coroides mediante técnicas de PCR: se encontraron varios productos de PCR obtenidos a partir de ARNm asociados a drusas (TIMP-3, apolipoproteína E, vitronectina, C3, C5, C9) en la neurorretina, el EPR, la coroides y/o células EPR humanas aisladas (Alexander et al, 1990; Anderson et al., 2001; Hageman et al., 1999; Mullins et al., 2000). También se han realizado estudios cuantitativos para identificar los niveles de expresión de las moléculas asociadas a las drusas con el fin de determinar si pueden expresarse localmente en cantidades significativas y si la expresión de los genes que codifican estas moléculas puede cambiar sustancialmente en función de la edad, el daño o la patología ocular. Por ejemplo, los análisis cuantitativos de PCR determinaron los niveles de expresión de tres proteínas asociadas a las drusas: apolipoproteína E, vitronectina y C5, en la retina, la EPR y la coroides. Las tasas estandarizadas de ARNm para la apolipoproteína E en la retina y la EPR/coroides en relación con el hígado fueron de 0,45 y 0,15, respectivamente, y las tasas de ARNm para la vitronectina en la retina y la EPR/coroides en relación con el hígado fueron de 0,47 y 0,06. Cabe señalar, por tanto, que los niveles de ARNm tanto para la apolipoproteína E como para la vitronectina fueron casi 50% de los medidos en el hígado, y significativamente superiores a las tasas en el cerebro en comparación con el hígado (0,28 y 0,01). Las tasas de ARNm C5 relacionadas con el hígado fueron de 0,45±0,55 y 0,14±0,12 en la retina y la EPR/coroides, respectivamente.
PAPEL DE LOS MACRÓFAGOS EN LA DMLE
Ambati et al. describen la degeneración de la retina en ratones que carecen de la proteína quimiotáctica de monocitos 1 (MCP-1), un factor quimiotáctico de macrófagos y células T de memoria, o de su ligando (CCR-2). La degeneración en estos ratones conduce al desarrollo de las formas neovascular (húmeda) y atrófica (seca) de la DMAE. Estos datos apoyan la hipótesis de que las células inflamatorias, en particular los macrófagos, pueden causar el daño en esta enfermedad.
Algunos consideran que las drusas de la DMAE son residuos de material no digerido procedente del mal funcionamiento de las células fagocíticas de la capa epitelial de la retina. Parecen consistir en "residuos" de diversos materiales biológicos: lípidos ricos en colesterol, diversas proteínas y más. En particular, se han identificado lipoproteínas oxidadas dentro de estas estructuras, lo que apoyaría la hipótesis de que podrían ser el resultado de un daño oxidativo. Las drusas también contienen sustancias potencialmente quimiotípicas hacia los macrófagos, como componentes del sistema del complemento e inmunoglobulinas.
Los estudios clínicos muestran una correlación entre la formación de drusas y la autofluorescencia (observada con un oftalmoscopio de barrido láser), una indicación indirecta de la acumulación de depósitos granulares en la capa celular del EPR. Estos depósitos, denominados gránulos de lipofuscina, son membranas celulares parcialmente degradadas dentro de lisosomas, y se acumulan en células que endocitan lipoproteínas oxidadas pero no pueden deshacerse de ellas adecuadamente. Esta deficiencia se produce tanto en células carroñeras predispuestas a esta tarea de eliminación de lipoproteínas, como los macrófagos envejecidos de las lesiones ateroscleróticas (células espumosas), como en fagocitos menos "profesionales", como las neuronas del sistema nervioso central y las células de la capa epitelial de la retina. Los defectos de esta función en las células epiteliales de la retina y los macrófagos de la coroides podrían contribuir a la acumulación de drusas con el paso del tiempo.
Además de su ya conocida actividad proinflamatoria, la MCP-1 también puede tener un papel inmunomodulador. Por ejemplo, parece favorecer el desplazamiento de la respuesta inmunitaria de las células T hacia las células T auxiliares de tipo II, promoviendo la producción de interleucina-4 (IL-4) y suprimiendo la citocina IL-12 asociada a la respuesta de las células T auxiliares de tipo I. Las células de la barrera hemato-retiniana, incluidas las células EPR, producen MCP-1 de forma constitutiva y pueden secretar grandes cantidades de la misma tras la exposición a otras citocinas y quimiocinas (como la IL-1B y el factor de necrosis tumoral alfa).
La experiencia de Ambati et al. respalda dicho papel homeostático e inmunorregulador de la MCP-1, así como su función quimiotáctica hacia los macrófagos proinflamatorios o proangiogénicos. La MCP-1 también podría coordinar el recambio de macrófagos residentes y células dendríticas inmaduras, ambos formando una red celular en la coroides, que está íntimamente en contacto con la capa de células epiteliales. Las células dendríticas inmaduras median en la tolerancia inmunológica y han sido implicadas en la tolerancia inducida a los antígenos. Además, la MCP-1 parece ser necesaria para la inducción de tolerancia inmunológica a antígenos derivados de la superficie de la mucosa, lo que podría realizarse mediante la inducción de tolerancia en las células dendríticas.
Podría tener sentido considerar que, en el ojo, una menor producción de MCP-1, por parte de las células senescentes y moribundas de la capa epitelial pigmentaria de la retina, podría afectar a la función homeostática y fagocítica de los macrófagos y células dendríticas residentes. En consecuencia, podrían acumularse drusas ricas en agentes quimiotácticos, con capacidad para iniciar una respuesta inflamatoria de bajo grado, que llevaría al reclutamiento de macrófagos proangiogénicos activados derivados de la médula ósea. Serían estos macrófagos los que podrían inducir la enfermedad degenerativa macular relacionada con la edad.
GENÉTICA Y DMLE
El gran avance en la historia de la inflamación se produjo hace aproximadamente un año, principalmente gracias a los polimorfismos de un solo nucleótido (SNP) humanos, sutiles cambios en la secuencia del ADN que pueden utilizarse para detectar variantes genéticas causantes de enfermedades. En los últimos años, los investigadores han utilizado los SNP para identificar diversas regiones cromosómicas que contienen genes que pueden influir en el riesgo de padecer DMAE. En marzo de 2006, tres equipos independientes dirigidos por Josephine Hoh, de Yale; Albert Edwards, de Dallas; y Lindsay Farrer, de Boston, informaron de lo que habían descubierto sobre un gen del cromosoma 1 que aumenta significativamente el riesgo de padecer DMAE. El gen codifica una proteína llamada Factor H del Complemento que mantiene el sistema del complemento bajo estricto control para que no ataque a las células sanas. Los investigadores descubrieron que las personas con una variante concreta del gen del Factor H eran más propensas a desarrollar DMAE. La variante de alto riesgo podría explicar hasta el 50% de los casos, presumiblemente porque el producto proteico del gen alterado es menos eficaz para inhibir la vía del complemento. Los genes relacionados con la degeneración macular asociada a la edad apoyan la noción de que la inflamación desempeña un papel central en el daño de la zona central de la retina en este trastorno de la visión. Hageman et al. informaron de un vínculo entre un gen y la activación de la cascada del complemento. Este gen, que se encuentra en el cromosoma 6, produce una proteína denominada factor B que interviene en la activación del complemento.
La infección puede ser un factor determinante en el desarrollo de la DMAE. Estudios realizados en ojos de pacientes con DMAE exudativa, caracterizada por el desarrollo de vasos sanguíneos en la mácula, mostraron pacientes con infección previa por Chlamydia. No obstante, es necesario confirmar una relación causal entre la infección por Chlamydia y la DMAE.
Pericak-Vance et al. también proponen la interacción entre el gen LOC387715 y el tabaquismo como uno de los factores ambientales de riesgo más importantes para la DMAE. Los investigadores descubrieron que el riesgo resultante de la asociación ¶ de fumar y ser portador de la variante del gen era mayor que la suma del riesgo de los dos factores individuales a la hora de promover el desarrollo de la DMAE.
El factor H y los genes B y LOC387715 no son los únicos factores genéticos que pueden influir en el riesgo de desarrollar DMAE. Por ejemplo, en un estudio publicado en enero, Pericak-Vance et al. se basaron en el análisis de ocho genes que resultaron estar implicados en el desarrollo de la DMAE. Su análisis incluía también el VEGF. El producto génico VEGF estimula el desarrollo de los vasos sanguíneos, lo que sugiere que puede estar implicado en la formación de la DMAE exudativa, que es la forma más grave. Pero los genes del complemento y el LOC387715 son sin duda los principales contribuyentes al riesgo de DMAE. Basándose en esta investigación, se puede entonces considerar seriamente el uso de medidas preventivas como evitar fumar, disminuir la ingesta de alimentos grasos y aumentar la ingesta de antioxidantes y carotenoides.
IDENTIFICACIÓN DEL FACTOR H
Hageman et al. afirman que una variación en el gen del factor H (HF1/CFH) aumenta drásticamente la probabilidad de desarrollar DMAE, así como glomerulonefritis membranosa-proliferativa de tipo II (GNMP II). HF1 codifica una proteína que interviene en la primera línea de defensa inmunológica del organismo (el sistema innato) contra las infecciones bacterianas y otros microbios. Existen variantes protectoras y de riesgo de este gen.
En estudios anteriores, Hageman, Mullins, Anderson y Johnson implicaron a la cascada del complemento, parte del sistema inmunológico innato, en la formación de drusas. Las drusas contienen residuos de EPR, procesos de células dendríticas y diversas moléculas asociadas al sistema inmunitario, como inmunoglobulinas, antígenos de clase II, diferentes componentes del complemento, activadores y reguladores. Uno de estos reguladores, el factor H, es un componente clave de la vía alternativa de activación del complemento. El conjunto de estas observaciones llevó a los investigadores a concluir que la DMAE, al igual que otras enfermedades seniles como la enfermedad de Alzheimer y la aterosclerosis, puede tener un gran componente inflamatorio implicado.
Los autores señalaron que la GNMP II, si se prescinde de su aparición precoz, tiene manifestaciones oculares indistinguibles de la DMAE. Una mutación puntual en el gen HF1 (I1166R) causa la GNMP II en cerdos y ratones con deficiencia grave del factor H, que desarrollan una gromeluronefritis grave. Además, los individuos afectados de dos grandes familias afectadas por MGPN III mostraron vinculación con el cromosoma 1q31-32, un locus próximo a la región 1q25-31 que anteriormente se había asociado a la DMAE. Todas estas observaciones llevaron a los investigadores a considerar el factor H como el principal candidato tanto para la AMDLE como para la MGPN II. La variante genética más frecuente de riesgo de desarrollar DMAE se asoció a casi la mitad de los 900 sujetos afectados del estudio, mientras que apareció en 29% de los 400 controles (P=10-3). La fuerza de esta asociación es mucho mayor en comparación con las anomalías genéticas anteriormente atribuidas a la DMAE (genes ABCA4, FBNL5, FBNL6, APOE).
La primera línea de defensa contra los microorganismos y otras partículas extrañas es el sistema del complemento, encargado de reconocer, atacar y eliminar los microorganismos invasores creando orificios en sus membranas. En algunos casos, sin embargo, la activación sostenida del complemento puede provocar una inflamación crónica, agravar el daño tisular local y contribuir significativamente a la progresión de la enfermedad, como ocurre en la enfermedad de Alzheimer y la aterosclerosis. Para evitar tales daños, ciertas proteínas, entre ellas el factor H, principal inhibidor soluble de la vía alternativa de activación del complemento, mantienen el sistema bajo estricto control. Dado que la mayoría de las variaciones identificadas ocupan lugares funcionales importantes de la proteína HF1, los investigadores sugirieron la posibilidad de que estas variantes de riesgo pudieran alterar el comportamiento de la proteína HF1 y obstaculizar su papel en la regulación de la vía del complemento en la respuesta inmunitaria.
Por tanto, podría argumentarse que los individuos con DMAE y GNMP II comparten un defecto funcional en la proteína del factor H que afecta a la funcionalidad del sistema del complemento. Las variaciones de un solo nucleótido podrían, por ejemplo, cambiar la unión del HF1 al fragmento C3b del complemento o a la proteína C reactiva, el ácido siálico o la heparina. Del mismo modo, estas variaciones podrían alterar la interacción entre el HF1 y los microbios, posiblemente haciendo que los tejidos, como el EPR, sean más susceptibles a la infección. Este concepto se basa en el hecho de que los activadores más importantes de la vía alternativa del complemento son moléculas que se encuentran fácilmente en muchas superficies bacterianas y víricas. La hipótesis final es que una disfunción del sistema del complemento puede provocar daños tisulares locales, especialmente en los lugares más vulnerables, como el glomérulo renal y la mácula. Hageman et al. también demostraron que la capa elástica de la membrana de Bruch es preferentemente más fina en la región macular. La alteración de esta fina capa y la inflamación subsiguiente podrían explicar la predilección de la mácula por la formación de lesiones, incluida la formación de membranas neovasculares coroideas.
El haplotipo HF1 de riesgo se asocia a una amplia gama fenotípica de DMAE, con la posible excepción de la atrofia geográfica, observada en un pequeño grupo de pacientes; por lo tanto, se supone que no existe una relación clara entre el genotipo asociado a la enfermedad y el fenotipo clínico.
Además, los resultados de este estudio son congruentes con ciertos factores de riesgo epidemiológicamente asociados a la DMAE. Por ejemplo, fumar inhibe la actividad del factor H y aumenta de 4 a 5 veces el riesgo de DMAE.
Los datos de Hageman et al. también concuerdan con afirmaciones anteriores sobre el papel del complemento en la DMAE. Parece probable que la herencia de un haplotipo HF1 de riesgo, en combinación con un agente infeccioso u otros activadores atípicos de la vía alternativa, como los complejos inmunes, el péptido beta amiloide o el colesterol, pueda aumentar sustancialmente la susceptibilidad individual a la DMAE y la GNMP II.
Está claro que las moléculas implicadas en la activación del complemento y su regulación serán futuras dianas para el desarrollo de pruebas de diagnóstico precoz y tratamientos terapéuticos para la DMAE y quizá también para otras enfermedades de origen inflamatorio.
EL GEN HTRA1
Un locus en el cromosoma 10q26 se ha relacionado desde hace tiempo con el riesgo de DMAE. Un polimorfismo de un solo nucleótido, rs11200638, en la región promotora del gen HTRA1, es la variante génica con mayor probabilidad de estar implicada en el desarrollo de la enfermedad, con un riesgo atribuible de 49,3%. Experimentos inmunohistoquímicos revelaron, mediante el uso de anticuerpos monoclonales, la presencia del producto HTRA1 en las drusas de los ojos con DMAE. El polimorfismo de nucleótido único rs11200638 se localiza 512 pb (pares de bases) aguas arriba del sitio de inicio de la transcripción del gen HTRA1. Este gen codifica para un miembro de la familia de las serina proteasas que se expresa en la retina y en la EPR (en ratones). HTRA1 parece regular la degradación de proteoglicanos en la matriz extracelular. Se cree que esta actividad facilita el acceso de las enzimas que degradan la matriz, como las colagenasas y las mateloproteasas de la matriz, a sus sustratos. La sobreexpresión de HTRA1 puede alterar la integridad de la membrana de Bruch, favoreciendo la invasión de los capilares coroideos a través de la matriz extracelular, como ocurre en la DMAE exudativa. Además, HTRA1 se une e inhibe el factor de crecimiento transformante ? (TGF-?), un importante regulador del depósito de matriz extracelular y de la angiogénesis. DeWan et al. informan de la misma asociación entre esta variación nucleotídica de HTRA1 y la DMAE exudativa en la población china.
TIMP-3 en la membrana de Bruch: cambios durante el envejecimiento y en la DMAE
Las metaloproteasas de matriz (MMP) y sus respectivos inhibidores tisulares (TIMP) desempeñan un papel importante en el recambio de la membrana extracelular (MEC). Las MMP constituyen una familia de enzimas secretoras, actualmente con más de 20 miembros, que participan en la degradación de los componentes de la MEC en el curso del recambio y la renovación normales. Las MMP también están implicadas en las primeras fases de la neovascularización, donde se cree que, junto con otras proteasas, pueden ser necesarias para la degradación de los componentes de la membrana capilar como requisito previo para la formación de nuevos vasos. Se cree que los TIMP, representados por cuatro productos génicos distintos, suprimen la degradación excesiva de la MEC y pueden desempeñar un papel funcional importante en la limitación de la neovascularización. El Timp-3, una vez secretado, se une a los componentes de la MEC, mientras que los demás TIMPs no lo hacen.
Una de las funciones de Timp-3 en la membrana de Bruch podría ser la de potente inhibidor local de la actividad de las MMP, regulando el recambio de la membrana de Bruch y limitando la neovascularización coroidea.
La distrofia del fondo de ojo de Sorsby es comparable a una degeneración macular hereditaria de aparición temprana, caracterizada por el engrosamiento de la membrana de Bruch y la neovascularización submacular. Se han encontrado mutaciones en el gen Timp-3 en familias con distrofia del fondo de ojo de Sorsby. Los estudios inmunohistoquímicos en un ojo donado de un paciente con distrofia de Sorsby mostraron una extensa acumulación de Timp-3 en la membrana de Bruch engrosada. Estas observaciones llevaron a evaluar la presencia de Timp-3 y su distribución en la membrana de Bruch engrosada de ojos con DMAE. Aunque hasta ahora no se han descubierto mutaciones en la región codificante o en los elementos reguladores del gen Timp-3 en pacientes con DMAE observados, el exceso de Timp-3 dentro de la MEC podría causar el engrosamiento de la membrana de Bruch e impedir la remodelación normal de la membrana. Debido a la importancia de la permeabilidad de la membrana de Bruch en el tráfico de metabolitos entre la coroides y el EPR, es importante comprender el papel de Timp-3 en el envejecimiento normal y en la DMAE.
El estudio dirigido por Motohiro Kamei y Joe G. Hollyfield analizó 36 ojos normales enucleados post mortem (entre 14-96 años) y 15 ojos con DMAE (entre 74-98 años) y evaluó el cambio en la distribución de TIMP-3 en relación con la edad. También se compararon los niveles de TIMP-3 en ojos de pacientes con DMAE y controles sanos de la misma edad.
El estudio demostró que el Timp-3 inmunorreactivo estaba presente en la membrana de Bruch en todas las muestras de tejido de donantes normales y se distribuía por todo el grosor de la membrana. Aunque la inmunorreactividad era uniforme en cada muestra de donante, en general, los ojos de los donantes más jóvenes mostraban menos inmunorreactividad que los ojos de los donantes de más edad. La inmunorreactividad de Timp-3 no era evidente en la retina neurosensorial, la coroides o la esclerótica.
Distribución y presencia de TIMP-3 en ojos con DMAE
Todos los ojos con DMLE utilizados contenían drusas blandas. Siempre que se observaron drusas blandas, ya fueran aisladas o confluentes, cada una de ellas era fuertemente inmunorreactiva con el anticuerpo anti-Timp-3. Las drusas duras, cuando estaban presentes, también eran fuertemente inmunorreactivas.
En la membrana de Bruch, bajo las extensas zonas de atrofia de la EPR, la inmunorreactividad de Timp-3 no era evidente o apenas detectable. La neovascularización coroidea estaba presente en 8 de 15 ojos de donantes con DMAE. En las zonas en las que el EPR había proliferado alrededor de una membrana neovascular coroidea o una cicatriz fibroblástica, el Timp-3 inmunorreactivo rodeaba al EPR hiperplásico. La inmunorreactividad era baja y mostraba una distribución irregular, probablemente debido a una pérdida de función y polaridad de la proliferación del EPR.
En las regiones de transición entre las zonas de atrofia de la RPE y una RPE normal, se observó una disminución de la inmunorreactividad de Timp-3, pero estaba presente en las drusas blandas y bajo la capa elástica central de la membrana de Bruch.
La cantidad de Timp-3 presente en la membrana de Bruch bajo la mácula en la retina humana normal parece depender de la edad.
Las secciones de la fóvea de donantes de la segunda a la tercera década de vida eran débilmente inmunorreactivas con el anticuerpo anti-TIMP-3. Además, la distribución de Timp-3 cambió en las décadas novena y décima, extendiéndose la inmunorreactividad desde la membrana de Bruch hasta la coriocapilar.
El análisis cuantitativo demostró que el contenido y la función de Timp-3 en la mácula aumentaban con la edad, con un coeficiente de correlación significativo (r = 0,66 y 0,67, respectivamente). Timp-3 es, por tanto, una proteína relacionada con la edad. El análisis cuantitativo indicó que los niveles de Timp-3 eran significativamente elevados en la mácula de los ojos con DMAE en comparación con los ojos normales.
Los ojos con DMLE, sin embargo, mostraron una distribución no uniforme, con una ausencia virtual de inmunorreactividad de Timp-3 en áreas de atrofia EPR y abundante inmunorreactividad en áreas fuera de las áreas atróficas donde la EPR estaba presente. Esto indica que la distribución de Timp-3 en la DMAE en ojos con atrofia del EPR no es uniforme.
Diversos factores hereditarios o no hereditarios, como alteraciones proteicas, estrés oxidativo o trastornos de enzimas hidrolíticas, pueden contribuir a la acumulación acelerada de estos residuos con la edad. Dado que el Timp-3 inhibe en gran medida las MMP, las drusas con un exceso de Timp-3 pueden retrasar la renovación de la membrana de Bruch. Esto puede provocar el engrosamiento de la membrana de Bruch, la reducción de la permeabilidad de la membrana de Bruch al tráfico de metabolitos y nutrientes entre la coroides y el EPR, y la posterior atrofia del EPR y los fotorreceptores.
En las zonas en las que se observó neovascularización coroidea, la RPE estaba ausente y no se apreciaba inmunorreactividad para Timp-3 en la membrana de Bruch subyacente.
Concluimos que el contenido de Timp-3 en la membrana de Bruch de la mácula aumenta durante el envejecimiento normal y que el contenido de Timp-3 se eleva por encima de los niveles normales en la región macular de los ojos con DMAE. Esto sugiere que Timp-3 puede ser una de las moléculas clave en el engrosamiento de la membrana de Bruch en el envejecimiento normal y en la DMAE.
Indicadores sistémicos de inflamación, disfunción endotelial y DMAE
Se ha planteado la hipótesis de que la inflamación desempeña un papel en la patogénesis de la DMAE. Hageman et al. indicaron que las drusas contienen proteínas relacionadas con procesos inmunomediados y con la inflamación. Se han observado células inflamatorias crónicas en la superficie externa de la membrana de Bruch en ojos con degeneración macular asociada a la edad de tipo exudativo. Estas células pueden causar lesiones aterogénicas y microvasculares por la liberación directa de oxidantes, compuestos tóxicos del oxígeno y enzimas proteolíticas que también pueden dañar la membrana de Bruch. Sin embargo, los datos de los estudios epidemiológicos observados hasta ahora no han indicado una relación consistente entre la inflamación sistémica o la presencia de marcadores inflamatorios y la DMAE.
La disfunción endotelial, derivada de la inflamación, la hipertensión, el tabaquismo y otros factores, se ha considerado un requisito previo en la patogénesis de la DMAE. El objetivo del estudio de Klein et al. era investigar si los marcadores de inflamación sistémica y disfunción endotelial estaban asociados a la DMAE. Los procedimientos seguidos en este estudio incluyeron: peso, talla y presión arterial sistémica, un cuestionario estandarizado, que incluía preguntas específicas sobre antecedentes de enfisema, artritis, tabaquismo, consumo de alcohol y uso de vitaminas.No se encontraron evidencias de la asociación de estos marcadores de inflamación sistémica con la DMAE, en concordancia con los datos del estudio cardiovascular (CHS). Esta falta de asociación entre los indicadores de inflamación sistémica estudiados (hsCRP sérica, SAA, Tnf-? e IL-6) y la DMAE coincide con la demostrada ineficacia del uso de antiinflamatorios sistémicos. En cambio, los ensayos clínicos han sugerido los posibles efectos beneficiosos de los corticosteroides administrados por vía intravítrea. Los corticosteroides intravítreos reducen la incidencia de membranas neovasculares inducidas por láser en primates al reducir la actividad de las células inflamatorias y su número en la coroides. Otros mecanismos potenciales incluyen la reducción de la expresión de VEGF y la regulación a la baja de la molécula de adhesión intracelular 1 (ICAM-1) expresada en las células EPR y el endotelio de los vasos, que regula la adhesión leucocitaria y la diapédesis durante la inflamación.
CONCLUSIONES
Cinco conceptos resumidos relevantes para la patología de la DMAE
En primer lugar, los cambios relacionados con la edad a los que se añaden acontecimientos patológicos conducen a la DMAE.
En segundo lugar, el estrés oxidativo a lo largo de los años y en la DMAE provoca daños en el EPR y la coriocapilar.
En tercer lugar, la EPR y el daño coriocapilar en la DMAE son el resultado de la inflamación crónica de la coroides y la membrana de Bruch.
En cuarto lugar, los daños en el EPR, la coriocapilar y la inflamación pueden provocar anomalías en la matriz extracelular (MEC). Esta matriz extracelular anómala provoca un deterioro de la difusión de nutrientes desde el EPR a la neurorretina, lo que puede provocar más daños en la retina y el EPR.
En quinto lugar, las anomalías de la matriz extracelular (MEC) provocan una alteración de la función del complejo EPR-coriocapilar que evoluciona hacia la atrofia de la retina, la atrofia del EPR y el crecimiento de nuevos vasos (NVC).
En esta secuencia de acontecimientos, el entorno y la genética pueden alterar el curso de la enfermedad.
Dra. Laura Bertazzi
Dr. Cristiano Luiselli
Dr. Pierfilippo Sabella
Cátedra de Oftalmología Clínica
Departamento de Ciencias Clínicas "Luigi Sacco" - Milán
Unidad Operativa de Oftalmología Hospital 'Luigi Sacco' - Milán
BIBLIOGRAFÍA
1. Klein R et al. Prevalence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology 99: 933-943, 1992.
2. Kahn HA et al. The Framingham Eye Study Outline and major prevalence findings. Am J Epidemiol 106: 17-32, 1977.
3. Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005 Jun;123(6):774-82.
4. Donoso LA, Kim D, Frost A, Callahan A, Hageman G. El papel de la inflamación en la patogénesis de la degeneración macular asociada a la edad. Surv Ophthalmol. 2006 Mar-Apr;51(2):137-52.
5. Zarbin MA. Conceptos actuales sobre la patogénesis de la degeneración macular asociada a la edad. Arch Ophthalmol. 2004 Abr;122(4):598-614.
6. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. Una hipótesis integrada que considera las drusas como biomarcadores de procesos inmunomediados en la interfaz EPR-membrana de Bruch en el envejecimiento y la degeneración macular asociada a la edad. Prog Retin Eye Res. 2001 Nov;20(6):705-32
7. Penfold PL, Killingsworth MC, Sarks SH. Degeneración macular senil: implicación de las células inmunocompetentes. Graefes Arch Clin Exp Ophthalmol. 1985;223(2):69-76.
8. Forrester JV. Los macrófagos en la degeneración macular. Nat Med 2003 Nov;9(11):1350-1
9. Ambati J, Anand A, Fernández S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. Un modelo animal de degeneración macular asociada a la edad en ratones senescentes deficientes en Ccl-2 o Ccr-2. Nat Med. 2003 Nov;9(11):1390-7.
10. Wiggs JL. Complement factor H and macular degeneration: the genome yields an important clue. Arch Ophtalmol. 2006 Apr;124(4):577-8.
11. Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, Boekhoorn SS, Vingerling JR, Hofman A, Oostra BA, Uitterlinden AG, Stijnen T, van Duijn CM, de Jong PT. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA. 2006 Jul 19;296(3):301-9.
12. Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA, Olson L, Kenealy SJ, Hauser M, Gilbert JR, Pericak-Vance MA. Genes candidatos funcionales en la degeneración macular asociada a la edad: asociación significativa con VEGF, VLDLR y LRP6. Invest Ophthalmol Vis Sci. 2006 Ene;47(1):329-35.
13. Emil Wirostko, William J. Wirostko, Barbara M. Wirostko. La degeneración macular asociada a la edad es una enfermedad inflamatoria posiblemente tratable con minociclinaActa Ophthalmol Scand. 2004 Abr;82(2):243-4.
14. Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. Una variante del gen HTRA1 aumenta la susceptibilidad a la degeneración macular asociada a la edad. Science. 2006 Nov 10;314(5801):992-3.
15. Kamei M, Hollyfield JG. TIMP-3 en la membrana de Bruch: cambios durante el envejecimiento y en la degeneración macular asociada a la edad. Invest Ophthalmol Vis Sci. 1999 Sep;40(10):2367-75.
16. Bailey TA, Alexander RA, Dubovy SR, Luthert PJ, Chong NH. Medición de la expresión de TIMP-3 y del grosor de la membrana de Bruch en la mácula humana. Exp Eye Res. 2001 Dec;73(6):851-8.
17. Strunnikova N, Hilmer S, Flippin J, Robinson M, Hoffman E, Csaky KG. Differences in gene expression profiles in dermal fibroblasts from control and patients with age-related macular degeneration elicited by oxidative injury. Free Radic Biol Med. 2005 Sep 15;39(6):781-96
18. Nakata K, Crabb JW, Hollyfield JG. Distribución de cristalinas en el complejo coroideo-membrana de Bruch de ojos con DMAE y ojos de donantes de la misma edad. Exp Eye Res. 2005 Jun;80(6):821-6.
19. Bok D. La evidencia de un proceso inflamatorio en la degeneración macular asociada a la edad gana nuevos apoyos. Proc Natl Acad Sci U S A. 2005 Mayo 17;102(20):7053-4.
20. Penfold PL, Killingsworth MC, Sarks SH. Degeneración macular senil. Implicación de las células gigantes en la atrofia del epitelio pigmentario de la retina. Invest Ophthalmol Vis Sci. 1986 Mar;27(3):364-71.
21. Penfold P, Killingsworth M, Sarks S. Estudio ultraestructural del papel de los leucocitos y los fibroblastos en la ruptura de la membrana de Bruch. Aust J Ophthalmol. 1984 Feb;12(1):23-31.
22. Vid AK, Stader J, Branham K, Musch DC, Swaroop ABiomarcadores de enfermedad cardiovascular como factores de riesgo de degeneración macular asociada a la edad. Oftalmología. 2005 Dic;112(12):2076-80.
Dr. Carmelo Chines
Director responsable