







La retinosis pigmentaria afecta a miles de personas en todo el mundo con una pérdida progresiva de visión que, en muchos casos, comienza en la infancia y sigue empeorando a lo largo de la vida. Es una enfermedad compleja, a menudo hereditaria, que puede afectar significativamente a la independencia y la calidad de vida. El mayor reto para la comunidad científica sigue siendo encontrar tratamientos que no sólo puedan ralentizar el deterioro visual, sino también proteger y preservar las células retinianas sanas.
Sin embargo, en los últimos años han surgido nuevas perspectivas alentadoras. Estudios recientes están revelando moléculas innovadoras capaces de estabilizar una proteína clave implicada en los procesos degenerativos de la retina. Este descubrimiento podría allanar el camino a terapias específicas revolucionarias capaces de intervenir directamente en los mecanismos biológicos que conducen a la pérdida de visión. Para muchas personas que padecen retinosis pigmentaria, estos resultados representan un rayo de esperanza en un futuro en el que por fin pueda controlarse la progresión de la enfermedad.
Descubra cómo la investigación está transformando la comprensión de esta enfermedad y qué nuevas posibilidades terapéuticas podrían hacerse realidad pronto.
¿Qué es la retinosis pigmentaria?
La retinosis pigmentaria es un trastorno genético que afecta a la retina, la membrana que recubre la parte posterointerna del globo ocular y se encarga de convertir la luz en señales eléctricas que el cerebro interpreta como imágenes. La enfermedad se desarrolla cuando los fotorreceptores -las células especializadas que nos permiten ver en la oscuridad (bastones) y distinguir colores y detalles (conos)- empiezan a degenerar.
Esta degeneración progresa lenta pero continuamente, provocando cambios en la percepción visual.
Aunque existen muchas formas y causas, el diagnóstico precoz permite seguir la evolución de la enfermedad y adoptar estrategias personalizadas para preservar al máximo la función ocular.
Síntomas y evolución de la enfermedad
Los síntomas de la retinosis pigmentaria pueden variar considerablemente de una persona a otra, dependiendo de la mutación genética implicada y de la edad de aparición. A pesar de esta variabilidad, algunos signos son comunes. Las primeras dificultades suelen afectar a la visión nocturna, a las que pueden seguir problemas para moverse en entornos poco iluminados o con fuertes contrastes de luz.
Otro síntoma característico es la pérdida gradual de la visión periférica: el campo visual tiende a estrecharse, creando finalmente una especie de "visión de túnel". Este cambio puede pasar desapercibido en las primeras fases, pero con el tiempo se hace más notable y limita significativamente la percepción de los obstáculos laterales.
Muchas personas notan que tropiezan con más facilidad o tienen problemas para moverse en entornos desconocidos.
La progresión puede ser lenta, durando años o décadas, o avanzar más rápidamente, haciendo que cada caso sea único en su curso.
En fases avanzadas, la visión central puede verse afectada, lo que dificulta actividades como leer, conducir o reconocer caras. Aunque actualmente no existe una cura definitiva, entender cómo evoluciona la enfermedad con el tiempo permite tomar medidas específicas para preservar la visión el mayor tiempo posible, utilizando ayudas visuales, tecnología de asistencia y revisiones periódicas.
La investigación sigue ofreciendo nuevas esperanzas, con terapias génicas, moléculas innovadoras y enfoques personalizados que podrían cambiar radicalmente la experiencia de las personas con retinosis pigmentaria.
Mutaciones genéticas y malformaciones de la rodopsina
El papel de las mutaciones genéticas en la retinosis pigmentaria es fundamental y uno de los campos más estudiados en los últimos años. Comprender cómo interfieren alteraciones genéticas específicas en el funcionamiento de la rodopsina, una proteína clave en el procesamiento visual, es esencial para desarrollar terapias más eficaces.
Mutaciones patogénicas relacionadas
Las mutaciones responsables de la retinosis pigmentaria suelen estar relacionadas con defectos genéticos hereditarios que afectan directamente a la estructura y función de la rodopsina. Esta proteína, presente en los bastones de la retina, es indispensable para que el ojo perciba la luz, especialmente en condiciones de poca luminosidad.
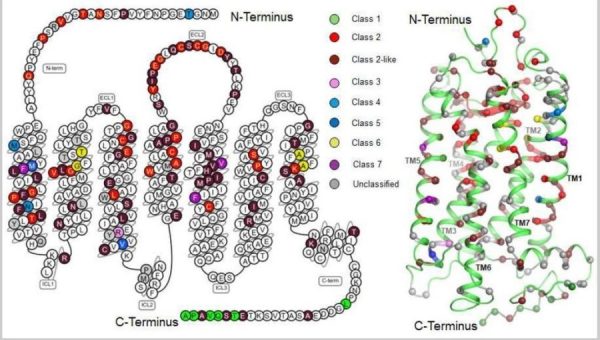
Cuando una mutación altera la estructura de la rodopsina, la proteína puede plegarse mal, volverse inestable o no alcanzar correctamente su posición en las células fotorreceptoras. Este mal funcionamiento desencadena una cascada de acontecimientos celulares que dañan gradualmente los fotorreceptores.
El mapeo genético de estas mutaciones se ha convertido en una herramienta fundamental, ya que permite identificar con precisión el origen del defecto, predecir la progresión del trastorno y, sobre todo, identificar dianas terapéuticas específicas. Conocer la mutación precisa también ayuda a orientar a los pacientes hacia ensayos clínicos y terapias personalizadas.
Efectos de las malformaciones en la vista
Las alteraciones de la rodopsina tienen efectos profundamente debilitantes sobre la visión, ya que merman la capacidad del ojo para convertir la luz en impulsos nerviosos. Sin una rodopsina funcional, los fotorreceptores entran en un estado de estrés constante, pierden funcionalidad y, con el tiempo, sufren la muerte celular.
Este proceso degenerativo da lugar a una pérdida progresiva de visión, que suele comenzar con dificultades en la visión nocturna y un estrechamiento del campo visual. A medida que los bastones degeneran, los conos -responsables de ver los colores y los detalles- también pueden resultar dañados, lo que conduce a un deterioro general de la capacidad visual.
Comprender cómo se produce esta degeneración es crucial porque permite a los investigadores desarrollar intervenciones dirigidas a estabilizar la rodopsina, proteger los fotorreceptores y ralentizar la progresión de la enfermedad.
Estrategias terapéuticas actuales
El tratamiento de la retinosis pigmentaria está evolucionando rápidamente, gracias a la llegada de tecnologías genéticas avanzadas y nuevas moléculas terapéuticas. Actualmente se están explorando tanto enfoques experimentales, como las terapias génicas, como estrategias clínicas para ayudar a los pacientes a convivir mejor con la enfermedad.
Nuevas terapias genéticas
Las terapias génicas representan una de las fronteras más prometedoras en el tratamiento de la retinosis pigmentaria. Estas intervenciones pretenden corregir directamente la mutación genética responsable de la disfunción de la rodopsina.
Utilizando vectores virales, los investigadores consiguieron introducir en la retina una copia corregida del gen, con el objetivo de restablecer la producción funcional de la proteína. Los primeros estudios clínicos han arrojado resultados alentadores: mejora de la sensibilidad a la luz y estabilización de la degeneración de los fotorreceptores.
Aunque la investigación sigue en curso y no se dispone de tratamientos para todas las variantes de la enfermedad, las terapias génicas abren la posibilidad de atajar la enfermedad antes de que aparezca, ofreciendo un enfoque que puede salvar la vida de muchos pacientes.
Opciones de tratamiento clínico para los pacientes
Junto a las terapias genéticas, existe una amplia gama de opciones clínicas y de apoyo para quienes padecen retinosis pigmentaria.
Estos enfoques incluyen:
- ayudas visuales avanzadascomo lentes con filtro, lupas digitales y dispositivos electrónicos que mejoran el contraste y facilitan la movilidad;
- apoyo psicológicoque es esencial para hacer frente al impacto emocional de la pérdida progresiva de visión;
- programas de rehabilitación visualque ayudan a los pacientes a mantener la autonomía en las actividades cotidianas.
Un equipo multidisciplinar -formado por oftalmólogos, genetistas, terapeutas visuales, psicólogos y asesores de rehabilitación- es esencial para crear un plan de tratamiento personalizado y poner al día al paciente sobre las nuevas opciones terapéuticas.
Nuevos avances en investigación
 La investigación científica atraviesa una fase de enorme progreso, gracias a técnicas computacionales avanzadas, modelos animales más precisos y nuevas moléculas diseñadas para estabilizar la rodopsina. Estos descubrimientos están cambiando rápidamente las perspectivas terapéuticas de la retinosis pigmentaria.
La investigación científica atraviesa una fase de enorme progreso, gracias a técnicas computacionales avanzadas, modelos animales más precisos y nuevas moléculas diseñadas para estabilizar la rodopsina. Estos descubrimientos están cambiando rápidamente las perspectivas terapéuticas de la retinosis pigmentaria.
Cribado virtual y moléculas correctoras
El cribado virtual es una de las herramientas más innovadoras utilizadas recientemente. Mediante algoritmos avanzados, los investigadores pueden simular miles de interacciones entre posibles moléculas terapéuticas y la rodopsina mutada, identificando las más prometedoras para corregir defectos estructurales.
Estudios recientes han identificado dos compuestos no retinoides con capacidad para atravesar la barrera hemato-retiniana y estabilizar la rodopsina malformada. Estas moléculas parecen impedir la degradación de los fotorreceptores, allanando el camino para nuevos tratamientos farmacológicos aplicables a distintas formas clínicas de retinosis pigmentaria relacionada con la rodopsina.
Impacto de los nuevos descubrimientos en los modelos animales
Los modelos animales siguen siendo un recurso inestimable para probar la eficacia de nuevas terapias. En ratones genéticamente predispuestos a desarrollar lesiones retinianas en respuesta a la luz brillante, se ha demostrado que las moléculas correctoras protegen la retina y prolongan la supervivencia de los fotorreceptores.
Estos resultados no sólo confirman el potencial terapéutico de las nuevas moléculas, sino que también ponen de relieve la posibilidad de aplicar estos enfoques a mayor escala. Si los datos se reproducen con éxito en humanos, podríamos asistir a un cambio radical en las estrategias de tratamiento de la retinosis pigmentaria.
Importancia del diagnóstico genético
El diagnóstico genético es una piedra angular en el tratamiento de la retinosis pigmentaria. Identificar la mutación responsable permite
- definir el pronóstico con mayor precisión,
- elegir estrategias terapéuticas específicas,
- participar en ensayos clínicos específicos,
- acceso de los familiares a programas de asesoramiento genético.
El diagnóstico precoz, especialmente en individuos con antecedentes familiares de la enfermedad, puede mejorar significativamente tanto el manejo clínico como las oportunidades de tratamiento.
Perspectivas de tratamiento para los pacientes
Las perspectivas de tratamiento de la retinosis pigmentaria están cambiando más rápido que nunca. Gracias al avance de las terapias génicas, al descubrimiento de moléculas correctoras y al desarrollo de tecnologías de asistencia cada vez más sofisticadas, los pacientes pueden esperar opciones cada vez más eficaces.
El objetivo, en los próximos años, es doble:
- preservar la vista el mayor tiempo posible,
- mejorar significativamente la calidad de vida.
Mantenerse informado sobre los avances científicos y mantener un diálogo continuo con los centros especializados es esencial para aprovechar al máximo los nuevos avances.
- Ortega JT, Gallagher JM, McKee AG, et al. Discovery of non-retinoid compounds that suppress the pathogenic effects of misfolded rhodopsin in a mouse model of retinitis pigmentosa. PLoS Biol. 2025 Jan 14;23(1):e3002932. doi: 10.1371/journal.pbio.3002932.
- Pashandi Z, Jastrzebska B. Small-Molecule Ligands of Rhodopsin and Their Therapeutic Potential in Retina Degeneration. Int J Mol Sci. 2025 Sep 15;26(18):8964. doi: 10.3390/ijms26188964.