








El retinoblastoma es una forma rara de tumor, que afecta a la retina y suele aparecer en niños antes de los 5 años, y está relacionado con mutaciones en el gen oncosupresor.
En todo el mundo, afecta a 1 de cada 15.000 niños y se calcula que cada año se diagnostican entre 8.600 y 9.000 nuevos casos. Por desgracia, alrededor del 90% de los niños con esta neoplasia viven en países en vías de desarrollo.
Aspectos genéticos
El retinoblastoma es una enfermedad de origen genético relacionada con mutaciones en el gen Rb1, localizado en el cromosoma 13. La proteína del retinoblastoma (pRb o Rb), de hecho, es una proteína supresora de tumores identificada en 1991, que se ha descubierto que no es funcional en varios tipos de cáncer. La función normal de Rb es bloquear la célula en una fase del ciclo celular, impidiendo que se divida de forma incorrecta o dañina. El retinoblastoma se desarrolla cuando ambos alelos del gen RB1, que codifica la proteína Rb, están inactivados por una mutación. El retinoblastoma puede ser transmitido por los padres en las formas hereditarias familiares (transmisión autosómica dominante) o puede surgir "ex novo" durante la embriogénesis en las formas hereditarias no familiares.
Diagnóstico
El diagnóstico del retinoblastoma es clínico, a través de ciertos signos típicos, en particular el reflejo blanquecino de las pupilas de los ojos afectados por la neoplasia. A menudo se descubre cuando se toma una fotografía del niño con flash y las pupilas aparecen blancas en lugar del reflejo rojizo-anaranjado normal, mostrando un característico reflejo blanquecino. Este signo se denomina "leucocoria" o "pupila de ojo de gato".
También puede provocar estrabismo o síntomas como enrojecimiento y dolor en los ojos. Esta última forma de pseudoinflamación se da en aproximadamente una décima parte de los pacientes, mientras que otros síntomas menos frecuentes son la protrusión del globo ocular, la decoloración del iris del ojo afectado, la presencia de diferencias en el tamaño, la forma y la actividad de la pupila, y otros síntomas relacionados con la metástasis, como el dolor de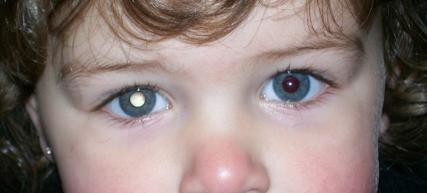 huesos. La agudeza visual puede verse afectada y el niño puede quejarse de visión doble.
huesos. La agudeza visual puede verse afectada y el niño puede quejarse de visión doble.
El diagnóstico debe confirmarse con un oftalmoscopia indirectadebe realizarse en midriasis y bajo anestesia en el caso de niños muy pequeños o lactantes. Debe examinarse cuidadosamente toda la cavidad ocular interna, toda la retina y la cabeza del nervio óptico.
A biomicroscopía con lámpara de hendiduramediante un potente haz de luz y un microscopio. Otras investigaciones pueden incluir fluorangiografía, ultrasonografía del globo ocular y resonancia magnética para obtener una imagen detallada del tejido blando con el fin de estadiar la extensión del tumor. La OCT, tomografía óptica computarizada, puede ser útil, pero es preferible evitarla en niños muy pequeños para no exponerlos al riesgo de exposición a radiaciones ionizantes, sobre todo si tienen antecedentes familiares de retinoblastoma y otros tumores.
Terapia
El enfoque terapéutico del retinoblastoma depende de la evaluación de la mayoría de estos factores, en concreto:
1. tamaño del tumor
2. número de localizaciones tumorales
3. localización del tumor
4. extensión y afectación de estructuras intraoculares y extraoculares
5. Presencia de otros tumores secundarios, ya que los tumores de pulmón o vejiga, los sarcomas u osteosarcomas y los melanomas son bastante frecuentes en los individuos con formas hereditarias de retinoblastoma.
6. antecedentes familiares de retinoblastoma
7. residuo visual conservado
Esta neoplasia ocular debe ser tratada por un equipo sanitario altamente especializado en el manejo de tumores pediátricos. El diagnóstico debe ser lo más precoz posible y el objetivo principal del tratamiento debe ser evitar la muerte por el tumor, salvar el ojo del niño
endolo inerte o en algunos casos atacable por otras técnicas ablativas.
y, en la medida de lo posible, garantizar la preservación de la función visual con los mínimos efectos secundarios.
Enfoques terapéuticos
A continuación se describen los enfoques terapéuticos utilizados actualmente.
1. Crioterapia
Se utiliza una criosonda para congelar y destruir tumores de hasta 5 mm de diámetro y 3 mm de grosor. Puede ser necesario repetir este tratamiento a intervalos de 3-4 semanas.
2. Fotocoagulación láser
La energía de la luz láser se utiliza para sobrecalentar el tumor y destruir la red vascular que lo alimenta, haciéndolo
3. Termoterapia
Las microondas se utilizan para calentar las células cancerosas y destruirlas.
4. Radioterapia
Se utilizan tanto la irradiación externa como la braquiterapia. La irradiación tiene la ventaja de una mayor precisión, que no afecta al tejido circundante y, por tanto, preserva mejor la visión. Actualmente se utilizan tres modalidades: IMRT (Radioterapia de Intensidad Modulada), Gamma Knife y la terapia de protones. La braquiterapia consiste en el uso de placas, en las que se colocan elementos radiactivos, que se suturan a la esclerótica en el tumor. La placa se deja colocada entre 3 y 7 días, dependiendo del tamaño y la extensión del tumor. La radioterapia conlleva graves riesgos de efectos secundarios en el cerebro y el ojo.
5. Quimioterapia: La quimioterapia sistémica se utiliza cuando se han producido metástasis a distancia. Los fármacos utilizados son el cisplatino, el etopósido y la vincristina. Puede administrarse por vía intravenosa, oral, arterial (a través de la arteria que irriga el globo ocular, sólo en el caso de tumores limitados al ojo) o intracraneal (en el espacio que rodea el sistema nervioso central y contiene líquido cefalorraquídeo).
6. Enucleación: Procedimiento quirúrgico que consiste en extirpar el ojo y parte del nervio óptico. Sólo se utiliza en los tumores más grandes y extensos, a menudo como tratamiento de segunda línea si la irradiación u otras terapias destinadas a salvar el ojo han fracasado, y sólo cuando no hay esperanza de salvar la vista. A menudo se asocia a la implantación de un ojo artificial del tamaño de un ojo adulto y que se vuelve a unir a los músculos oculares para garantizar unos movimientos más naturales. En este caso, los niños deben ser vigilados durante al menos dos años para detectar cualquier recidiva.
7. Exenteratio Orbitae: Extirpación del contenido de la órbita, incluidos los anexos oculares, es decir, el globo ocular, la musculatura extrínseca, la grasa peribulbar, la periorbita, los párpados y, posiblemente, la piel periocular. Representa el procedimiento más invasivo y destructivo.
Supervivencia
Las tasas de incidencia y supervivencia varían sustancialmente de un país a otro y están relacionadas con la precocidad del diagnóstico. Un estudio epidemiológico que acaba de publicarse en Jama Ophthalmology, Grupo de trabajo EUROCARE-6. Supervivencia y carga asistencial de los niños con retinoblastoma en Europa. analizaron datos sobre niños diagnosticados de retinoblastoma, recogidos en 81 registros de cáncer de 31 países europeos participantes en el proyecto EUROCARE-6, que abarcan los 14 años comprendidos entre enero de 2000 y diciembre de 2013. La incidencia del retinoblastoma en Europa se ha mantenido estable en 4 casos por millón de niños de 0 a 14 años, con una esperanza de vida a los 5 años de 97,8% En todo el mundo, una media de 9 de cada 10 niños diagnosticados de retinoblastoma pueden curarse y no presentan recidiva tumoral en los 5 años siguientes al tratamiento. Es necesario un seguimiento de por vida debido al alto riesgo de tumores secundarios o de desarrollar un tumor en el ojo contralateral. Este último suele aparecer en los 3 primeros años tras la aparición del primer tumor, por lo que los ojos deben examinarse cada 2-4 meses durante al menos los primeros 28 meses.
Sobre el tema de la oftalmología pediátrica, véase también
- COVID-19: efectos a largo plazo en niños - Oculista Italiano
- La "salud digital" de niños y jóvenes - Oculista Italiano
- Traumatismos retinianos en niños: cuidado con el balón - Oculista Italiano
- Insuficiencia de convergencia en niños: cómo intervenir - Oculista Italiano
- Chévez-Barrios P, Chantada GL, Wilson MW. Incidencia y tasas de supervivencia en niños europeos con retinoblastoma. JAMA Ophthalmol. 2024;142(11):1071–1072. doi:10.1001/jamaophthalmol.2024.4268
- Virgili G, Capocaccia R, Botta L, et al; Grupo de trabajo EUROCARE-6. Supervivencia y carga asistencial de los niños con retinoblastoma en Europa. JAMA Ophthalmol. Publicado en línea el 10 de octubre de 2024. doi:10.1001/jamaophthalmol.2024.4140