







La Degenerazione Maculare senile (DMLE) è la causa principale di grave riduzione dell’acuità visiva nelle persone oltre i 60 anni nel mondo occidentale. È una complessa patologia che riconosce diversi fattori di rischio come età, sesso, razza, esposizione alla luce, dieta, fumo di sigaretta e patologie vascolari.
Recentemente altri elementi sembrano giocare un ruolo importante nella patogenesi della malattia: variazioni di sequenze di DNA codificanti per i fattori del complemento, modificazioni del TMP-3 e attivazione di citochine infiammmatorie. La conoscenza di nuovi fattori coinvolti nella patogenesi di questa malattia potrà in futuro aprire la strada a nuove strategie terapeutiche per combattere la DMLE.
INTRODUZIONE
La Degenerazione Maculare legata all’età (DMLE) è un’affezione degenerativa che coinvolge la regione maculare in persone di età superiore ai 55 anni. La gravità e la frequenza ne fanno la causa principale di grave riduzione dell’acuità visiva, definita “cecità legale”, nelle persone oltre i 60 anni nel mondo occidentale. La DMLE può essere divisa clinicamente in una forma asciutta (dry), la più frequente e meno grave, ed in una essudativa (wet). Quest’ultima rappresenta il 20% circa della DMLE, ma è la causa di circa il 90% dei gravi casi di riduzione irreversibile dell’acuità visiva, sempre legati a questa patologia. Tale malattia comporta una progressiva perdita della porzione centrale del campo visivo, con risparmio di quella periferica, il che vuol dire non poter più leggere, guidare, riconoscere i volti delle persone, il numero dell’autobus, ecc.
Secondo stime abbastanza recenti, ogni anno nel mondo compaiono circa 500.000 nuovi casi di DMLE essudativa e questo numero, con l’aumento della vita media, tenderà a salire vertiginosamente nei prossimi decenni. Si stima che 8,5 milioni di americani siano attualmente affetti da tale patologia e, secondo le previsioni, entro il 2020 si prevedono circa 7,5 milioni di nuovi casi, sempre negli Stati Uniti.
La degenerazione maculare senile è una malattia complessa da studiare a causa del potenziale coinvolgimento di vari fattori tra cui quello demografico, ambientale, i rischi legati all’età, sesso, razza, esposizione alla luce, dieta, fumo e malattie cardiovascolari.
Le degenerazioni maculari e/o le distrofie maculari possono essere classificate in forme giovanili e forme senili. Per quanto riguarda la genetica molecolare delle distrofie maculari giovanili, la ricerca durante gli ultimi 10 anni ha contribuito significativamente alla comprensione di queste forme. Sono state identificate le caratteristiche cliniche e i geni coinvolti. Gran parte dei progressi nella comprensione delle distrofie maculari giovanili sono correlati all’esordio precoce della patologia. Collettivamente, questi studi hanno condotto all’identificazione dei numerosi geni addetti al processo visivo.
Recentemente si è scoperto che polimorfismi di singoli nucleotidi e variazioni di sequenza del DNA, riscontrati all’interno del gene del fattore H del complemento (CFH), sono fortemente associati con lo sviluppo di DMLE nei caucasici. Gli stessi autori hanno riscontrato l’implicazione di altri geni nella DMLE, tra i quali il gene VEGF e due geni coinvolti nel metabolismo lipidico. Il polimorfismo di un singolo nucleotide di CFH, Tyr402His, è stato associato con circa il 50% dei casi di DMLE. Un altro gene coinvolto nella cascata del complemento è stato associato alla patogenesi della DMLE. Questo gene, localizzato sul cromosoma 6, produce una proteina denominata Fattore B, che attiva il complemento. Un terzo gene coinvolto (LOC387715) è stato mappato sul cromosoma 10; il rischio di sviluppo di DMLE sembra essere correlato alla variazione di un singolo aminoacido nella proteina codificata, estranea alla cascata del complemento. Le analisi genetiche indicano che l’effetto di LOC387715 è indipendente da quello del fattore H, ma altrettanto importante, forse contribuendo a quasi il 40% dei casi di DMLE. Pericak-Vance et al. hanno evidenziato una relazione tra la variante ad alto rischio di LOC387715 e il fumo di sigaretta, uno dei maggiori fattori di rischio ambientali per DMLE. I dati fin qui riportati mostrano quanto sia importante che la ricerca scientifica possa, in termini di prevenzione, sviluppare una nuova terapia in grado di arrestare la drammatica evoluzione di questa patologia realmente invalidante.
Per quanto riguarda la DMLE asciutta esistono diversi studi:
· The Age-related Eye Disease Study (AREDS):
Trial multicentrico statunitense allestito per valutare se vitamine, antiossidanti e/o minerali riducano lo sviluppo e/o la progressione dalla malattia.
· The Prophylactic Treatment of Age-related Macular Degeneration (PTAMPD) Clinical Trial:
Studio pilota randomizzato sulla efficacia del trattamento laser delle soft drusen al polo posteriore che ha dimostrato, a due anni, una riduzione delle drusen nel 70% dei soggetti trattati e, di conseguenza, un miglioramento dell’acuità visiva. Per la DMLE essudativa, al trattamento laser tradizionale basato sulla distruzione delle aree neovascolari, con conseguente danno anatomico e funzionale della retina trattata, si sono aggiunte nuove proposte:
· Trattamento focale dei “feeder vessels”: basato sulla fotocoagulazione a bassa intensità con argon-laser e/o laser a diodi (infrarosso) dei neovasi afferenti (feeder vessels) delle membrane neovascolari, identificati grazie all’angiografia dinamica con fluoresceina sodica e verde di indocianina con oftalmoscopi a scansione laser.
· Terapia Termica Transpupillare (TTT): è indicata soprattutto per le forme “occulte”, sfrutta l’effetto termico del laser a diodi per provocare l’occlusione delle membrane neovascolari senza distruggere la retina sovrastante. È comunque una tecnica che necessita ancora di ulteriori studi e modifiche; la difficoltà di tale tecnica è legata alla mancanza di dati sulla corretta dose da utilizzare. È stata introdotta per il trattamento dei piccoli melanomi della coroide del polo posteriore.
· Asportazione chirurgica: i risultati sono abbastanza deludenti nella DMLE per i danni iatrogeni sull’epitelio pigmentato retinico. Migliori sono i risultati con la “rotazione” della retina (Machemer, Erkhardt e Todt) e la “traslocazione limitata” (De Juan, Tano), che sposta la regione foveale dalla sottostante membrana, in modo che quest’ultima possa essere fotocoagulata.
· Terapia radiante: si propone di prevenire la proliferazione di cellule endoteliali dei neovasi coroideali ed indurne la chiusura mediante radiazioni ionizzanti. Tuttavia studi clinici randomizzati (RAD) non hanno dato buoni risultati.
· Terapia fotodinamica (PDT): prima del suo avvento erano scarse le possibilità di intervento per le neovascolarizzazioni coroideali subfoveali. Tre trial multicentrici, TAP, VIP e VIT (quest’ultimo svoltosi in Italia e coordinato dalla Clinica Oculistica dell’Ospedale San Paolo di Milano) ne hanno dimostrato l’efficacia.
· Terapia farmacologica: trattasi di farmaci cortisonici (Triamcinolone Acetonide) e farmaci ad azione antiangiogenetica e angiostatica come gli anti-VEGF AVASTIN, MACUGEN e LUCENTIS, somministrati tutti intravitrealmente.
CARATTERISTICHE CLINICHE
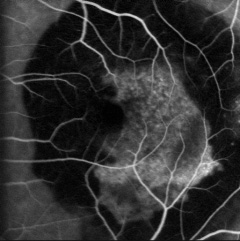
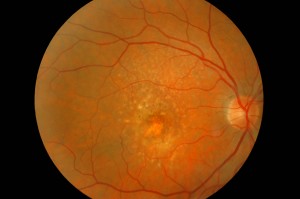
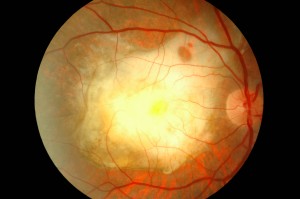
La DMLE, come già accennato, è stata classificata in due condizioni cliniche: una forma secca ed una umida. La neovascolarizzazione coroideale è caratteristica della forma umida, una fase che si ritrova nel 10% circa dei casi. Sebbene entrambe le forme di DMLE possano causare perdita visiva, la forma umida è coinvolta nel 90% circa dei casi di perdita visiva severa. È, inoltre, generalmente accettato che la forma umida della DMLE è preceduta e si sviluppa dalla forma secca. Al contrario la forma secca è molto più comune ed è caratterizzata clinicamente dalla presenza di drusen maculari, che sono depositi localizzati fra l’EPR e la membrana di Bruch, e da atrofia geografica, caratterizzata dalla morte delle cellule dell’EPR con atrofia dei fotorecettori sovrastanti.
La DMLE solitamente non è clinicamente evidente prima dei 50 anni. Dai reperti autoptici, la DMLE è riscontrata nel 33% degli occhi delle persone con più di 65 anni e nel 30% circa delle persone al di sopra dei 75 anni. La prevalenza della malattia varia dall’1,6% in soggetti di età compresa tra i 52 e i 64 anni al 27,9% in quelli oltre i 75 anni. La prevalenza di DMLE è trascurabile all’età di 50 anni raggiunge il 6% a 80 anni.
Tali discrepanze possono essere spiegate, in parte, per il fatto che l’occhio, così come altri organi, subisce un processo normale di invecchiamento indipendente dalla DMLE che, diversamente dalle distrofie maculari giovanili, può rendere la diagnosi più difficile. Inoltre la DMLE è associata a malattie cardiovascolari e ad altri disordini importanti, per cui è possibile che molti soggetti potenzialmente affetti possano decedere a causa delle patologie cardiovascolari, prima che la DMLE si manifesti, così che la prevalenza della patologia negli 80enni si abbassi.
FATTORI DI RISCHIO DEMOGRAFICI ED AMBIENTALI PER LA DMLE
L’età, il sesso, la razza, l’esposizione alla luce, patologie cardiovascolari concomitanti, la dieta, il fumo sono possibili fattori di rischio per la DMLE. La DMLE ha una prevalenza maggiore nella popolazione bianca, rispetto ai non-bianchi. Esistono studi contrastanti sull’effetto della esposizione cronica alla luce (ultravioletta o visibile) sulla retina.
LE DRUSEN COME MARKERS IMMUNOLOGICI DI DMLE
I costituenti molecolari e cellulari delle drusen sono stati analizzati approfonditamente. Gran parte del materiale ritrovato nelle drusen è sintetizzato da cellule che normalmente si trovano nell’occhio, ma parte del materiale deriva invece da fonti extraoculari. Per esempio il complemento, i lipidi, le lipoproteine B ed E sono costituenti comuni delle drusen oculari e delle placche aterosclerotiche, suggerendo che gli stessi processi biochimici ed immunologici possano essere coinvolti in entrambe le condizioni. Per esempio l’amiloide beta, una delle componenti infiammatorie principali delle placche nell’Alzheimer, si ritrova anche nelle drusen. Hageman et al. hanno proposto che le drusen possano essere il prodotto di una risposta infiammatoria localizzata che segue un danno dell’EPR coinvolgendo gli antigeni HLA ed il sistema del complemento. L’ipotesi si basa sull’osservazione delle drusen nell’MPGNII, una patologia renale in cui una disfunzione immunologica mediata dal complemento conduce a insufficienza renale. In questa condizione le drusen sono identiche a quelle della DMLE.
Le drusen sono depositi focali di materiale extracellulare, siti tra la membrana basale dell’EPR e lo strato collagene della membrana di Bruch. Le drusen sono importanti fattori di rischio e indicatori biologici della degenerazione maculare senile.
Sono comunemente osservate negli individui di età maggiore di 60 anni e nel contesto clinico della DMLE. Le dimensioni, il numero, l’estensione e la confluenza delle drusen sono importanti determinanti nel rischio di sviluppo di DMLE (Pauleikhoff et al., 1990). La presenza di drusen soft, grandi e/o confluenti è correlata alla comparsa di neovascolarizzazione coroideale, con un rischio relativo di 2,1 negli occhi con cinque o più drusen e di 1,5 negli occhi con una o più grosse drusen (Macular Photocoagulation Study Group, 1993).
La presenza di drusen è associata con vari deficit visivi che si manifestano prima della perdita dell’acuità visiva: cambiamenti nella sensibilità ai colori e al contrasto, nel recupero funzionale maculare, nella sensibilità visiva centrale, nella sensibilità al contrasto spaziotemporale (Frennesson et al., 1995; Holz et al., 1995; Midena et al., 1997, 1994; Stangos et al., 1995; Tolentino et al., 1994).
Diversi studi sono stati intrapresi nell’intento di determinare la composizione delle drusen, nella convinzione che comprendere la costituzione di un deposito correlato a una patologia potesse apportare nuove informazioni sul processo patogenetico stesso.
Lipidi
Donders per primo nel 1854 osservò la presenza di lipidi nelle drusen. Con l’uso di tecniche istochimiche ed enzimatiche si concluse che la componente lipidica delle drusen comprendeva probabilmente molecole glicolipidiche, cerebrosidi e/o gangliosidi (Wolter and Falls, 1962; Farkas et al., 1971; Pauleikhoff et al., 1992). Pauleikhoff et al. (1992) suggerirono l’esistenza di classi distinte di drusen, alcune con caratteristiche più “idrofile”, altre più “idrofobe”. Holz et al. (1994) hanno riscontrato una maggiore concentrazione lipidica nella regione maculare rispetto alle regioni periferiche della membrana di Bruch; le caratteristiche di questi lipidi farebbero supporre una loro origine cellulare piuttosto che vascolare. Al contrario Curcio et al. (2001) descrivono la presenza di entrambe le forme di colesterolo esterificato e non esterificato nella membrana di Bruch e nelle drusen: tali autori ipotizzano che la quantità notevole di colesterolo esterificato nelle drusen possa implicare una fonte vascolare (plasmatica) dei lipidi associati alle drusen e alla membrana di Bruch. Studi condotti da Haimovici et al. (2001) comprovano la presenza di esteri di colesterolo nelle drusen.
Nonostante i numerosi studi condotti, la fonte (cellulare, vascolare o combinata) dei lipidi associati alle drusen rimane un enigma. Tuttavia Hageman et al. hanno appurato che i geni per alcune lipoproteine associate alle drusen (come la lipoproteina E) sono trascritti a livello locale, mentre altre molecole (come la componente P dell’amiloide) sembra avere un’origine epatica, e arriverebbe alla membrana di Bruch tramite il circolo. Anche i lipidi associati alle drusen potrebbero avere allo stesso modo un’origine eterogenea.
Carboidrati
Farkas et al. per primi ammisero l’esistenza di glicolipidi all’interno delle drusen (1971). Kliffen et al. (1994; 1996), Farkas et al. (1971; 1996) identificarono glicoconiugati ricchi in glicosaminoglicani in depositi all’interno della membrana di Bruch.
Mullins et al. (1997) identificarono, come carboidrati maggiormente rappresentati nelle drusen, sia soffici che dure, glicocomposti con terminazioni costituite da glucosio/mannosio, N-acetilglucosamina, acido sialico.
Quando si provò a rimuovere, tramite neuraminidasi, i residui di acido sialico delle terminazioni dei carboidrati associati alle drusen, fu possibile ritrovare disaccaridi di galattosamina (Mullins e Hageman, 1999). Questi disaccaridi erano confinati in un core centrale costituito da glicoproteine con carboidrati legati tramite legami O-glicosidici. La presenza di questi domini costituenti un core all’interno delle drusen è indicativa di un ruolo del core nei primissimi stadi dello sviluppo delle drusen, simile all’accrescimento di una perla nella conchiglia di un’ostrica.
Proteine
I costituenti proteici delle drusen identificati per primi con metodi immunoistochimici includevano ubiquitina (Loeffler e Mangini, 1997), integrine (Brem et al., 1994), inibitori tissutali delle metalloproteasi (Fariss et al., 1997), prodotti finali della glicosilazione (Ishibashi et al., 1998), beta-amiloide (Loeffler et al., 1995), fibronectina (Pauleikhoff et al., 1992) e C1q (van der Schaft et al., 1993). Negli ultimi anni sono state identificate la vitronectina (Hageman et al., 1999), la componente P dell’amiloide, l’apolipoproteina E, il fattore X, le catene lambda delle immunoglobuline, fattori di attivazione del complemento, come il complesso C5b-9, e antigeni MHC di classe II (Johnson et al., 2000; Mullins et al., 2000; Mullins e Hageman, 1997). L’eterogeneità molecolare non corrisponde a dei fenotipi ultrastrutturalmente o clinicamente definiti (Hageman e Mullins, 1999).
Virtualmente tutte le proteine recentemente scoperte nelle drusen sono in qualche modo associate all’infiammazione e/o ad altri processi immuno-associati. Alcune sono classiche proteine di fase acuta, mentre altre sono componenti della cascata del complemento, o inibitori della via di attacco alla membrana del complemento. Altri ancora sono associati con l’attivazione immunologica, la coagulazione, la fibrinolisi. Inoltre molte di queste molecole sono comuni nei depositi patologici associati ad altre malattie come il morbo di Alzheimer, l’aterosclerosi, l’elastosi, l’amiloidosi, le glomerulonefriti (Mullins et al., 2000): così si è pensato fosse possibile che percorsi patogenetici comuni potessero essere coinvolti nella loro formazione.
Componenti cellulari
Secondo i modelli tradizionali della formazione delle drusen, qualsiasi materiale cellulare al loro interno era di origine dall’EPR. Infatti nelle drusen “iniziali” si possono ritrovare frammenti cellulari e organelli derivati dall’EPR, addirittura intere cellule. Inoltre sono state descritte vescicole di EPR che si estendono attraverso la membrana basale dell’EPR all’interno delle drusen o nei siti della loro successiva formazione (Ishibashi et al., 1986). Costituenti dell’EPR, come lipofuscina e melanina, si possono talvolta osservare nelle piccole e precoci drusen.
Un’osservazione nuova e potenzialmente molto significativa consiste nel fatto che molecole normalmente associate alle cellule, inclusi gli antigeni HLA-DR e CD specifici, si associano alle drusen. Queste molecole sono spesso localizzate in domini solitari, tipo “core”, nel contesto delle drusen (Mullins et al., 2000). Analisi immunofenotipiche recenti hanno documentato che questi core derivano da estensioni cellulari di cellule dendritiche coroideali, potenti antigen presenting cells associate con svariati processi di immunomodulazione. Inoltre questi processi delle cellule dendritiche sono tipicamente associati alle vescicole di EPR già descritte. Questi dati suggeriscono per la prima volta che il processo di formazione delle drusen possa essere cellulo-mediato e che specifiche vie immunomediate possano avere dei ruoli significativi in questi eventi.
mRNA
Molte delle molecole associate alle drusen, identificate recentemente usando metodi immunoistochimici, sono sintetizzate principalmente nel fegato. La loro fuoriuscita dai capillari della coroide e la conseguente aggregazione lungo la membrana di Bruch costituisce un potenziale avvio verso la loro deposizione all’interno delle drusen. Ad ogni modo alcuni tipi cellulari locali nella neuroretina, nell’EPR e/o nella coroide, che si trovano in prossimità della membrana di Bruch potrebbero anch’essi avere la capacità di sintetizzare parte di queste molecole. Di recente è stata ottenuta la prova della trascrizione genica di alcune molecole associate alle drusen nella neuroretina, nell’EPR e nella coroide con l’uso di tecniche di PCR: diversi prodotti di PCR ottenuti a partire da mRNA associati alle drusen (TIMP-3, apolipoproteina E, vitronectina, C3, C5, C9) sono stati rinvenuti nella neuroretina, nell’EPR, nella coroide e/o in cellule dell’EPR umane isolate (Alexander et al., 1990; Anderson et al., 2001; Hageman et al., 1999; Mullins et al., 2000). Sono stati anche intrapresi studi di tipo quantitativo per identificare i livelli di espressione delle molecole associate alle drusen, al fine di determinare se possano essere espresse localmente in quantità significative e se l’espressione dei geni codificanti per tali molecole possa cambiare sostanzialmente in funzione dell’età, del danno, di una patologia oculare. Per esempio, le analisi PCR di tipo quantitativo hanno determinato i livelli di espressione per tre proteine associate alle drusen: apolipoproteina E, vitronectina e C5, nella retina, nell’EPR e nella coroide. I tassi standardizzati dell’mRNA per l’apolipoproteina E nella retina e nell’EPR/coroide in relazione al fegato erano rispettivamente 0,45 e 0,15, e i tassi di mRNA per la vitronectina nella retina e nell’EPR/coroide rispetto al fegato erano 0,47 e 0,06. Da sottolineare, quindi, che i livelli di mRNA per entrambi l’apolipoproteina E e la vitronectina erano quasi il 50% di quelli misurati nel fegato, e significativamente più alti dei tassi nel cervello rispetto al fegato (0.28 e 0.01). I tassi dell’mRNA di C5 in relazione al fegato erano 0.45±0.55 e 0.14±0.12 nella retina e nell’EPR/coroide rispettivamente.
RUOLO DEI MACROFAGI NELLA DMLE
Ambati et al. descrivono la degenerazione retinica in topi che mancano della proteina chemotattica 1 dei monociti (MCP-1), un fattore chemotattico dei macrofagi e delle cellule T di memoria, o del suo ligando (CCR-2). La degenerazione in questi topi porta allo sviluppo di entrambe le forme neovascolare (umida) e atrofica (secca) della DMLE. Questi dati sostengono l’ipotesi che cellule infiammatorie, in particolare i macrofagi, possano essere causa del danno in questa patologia.
Le drusen della DMLE sono considerate da alcuni il residuo di materiale non digerito proveniente da cellule fagocitiche mal funzionanti dello strato epiteliale retinico. Sembrerebbero consistere in “scarti” di vario materiale biologico: lipidi ricchi di colesterolo, svariate proteine ed altro. In particolare, all’interno di queste strutture sono state identificate lipoproteine ossidate, il che sosterrebbe l’ipotesi secondo la quale potrebbero derivare da un danno ossidativo. Le drusen contengono anche sostanze potenzialmente chemotattiche verso i macrofagi, come componenti del sistema del complemento e immunoglobuline.
Studi clinici mostrano una correlazione tra la formazione delle drusen e l’autofluorescenza (osservata tramite un oftalmoscopio scanning laser), indice indiretto dell’accumulo di depositi granulari nello strato cellulare dell’EPR. Questi depositi, denominati granuli di lipofuscina, sono membrane cellulari parzialmente degradate all’interno di lisosomi, e si accumulano in cellule che endocitano lipoproteine ossidate, ma che non possono disporne in modo adeguato. Questa deficienza si verifica sia in cellule scavenger predisposte a questo compito di smaltimento delle lipoproteine, come i macrofagi invecchiati nelle lesioni aterosclerotiche (cellule schiumose), sia in fagociti meno “professionali”, come i neuroni del sistema nervoso centrale e le cellule dello strato epiteliale retinico. Difetti di tale funzione nelle cellule epiteliali retiniche e nei macrofagi della coroide potrebbero contribuire all’accumulo di drusen nel tempo.
Oltre alla già conosciuta attività proinfiammatoria, MCP-1 potrebbe anche possedere un ruolo immunomodulatorio. Per esempio sembra favorire lo shift della risposta immune T-cellulare verso i T-helper di tipo II, promuovendo la produzione dell’interleuchina-4 (IL-4) e sopprimendo la citochina IL-12 associata alla risposta T-helper di tipo I. Le cellule della barriera emato-retinica, comprese quelle dell’EPR, producono costitutivamente MCP-1 e possono secernerne in grande quantità dopo l’esposizione ad altre citochine e chemochine (quali IL-1B e il fattore di necrosi tumorale alfa).
L’esperienza di Ambati et al. supporta tale ruolo omeostatico e di immunoregolazione di MCP-1, così come la sua funzione chemotattica verso i macrofagi proinfiammatori o proangiogenici. MCP-1 potrebbe anche coordinare il turnover dei macrofagi residenti e delle cellule dendritiche immature, che formano entrambi una rete cellulare nella coroide, che è intimamente a contatto con lo strato cellulare epiteliale. Cellule dendritiche immature mediano la tolleranza immunologica e sono state implicate nella tolleranza indotta agli antigeni. Inoltre MCP-1 sembra essere richiesto per l’induzione della tolleranza immunologica verso gli antigeni che derivano dalla superficie mucosa, che potrebbe realizzare tramite l’induzione della tolleranza in cellule dendritiche.
Potrebbe avere senso la considerazione che, nell’occhio, una diminuita produzione di MCP-1, da parte di cellule senescenti e morenti dello strato epiteliale retinico, possa intaccare la funzione omeostatica e fagocitica dei macrofagi e delle cellule dendritiche residenti. Di conseguenza potrebbero accumularsi drusen ricche di agenti chemotattici, con la capacità di iniziare una risposta infiammatoria di basso grado, che condurrebbe al reclutamento di macrofagi proangiogenici attivati derivati dal midollo osseo. Sarebbero questi macrofagi che potrebbero indurre la patologia degenerativa maculare senile.
GENETICA E DMLE
La svolta nella storia dell’infiammazione si è avuta circa un anno fa, principalmente grazie ai polimorfismi umani di singoli-nucleotidi (SNPs), cambiamenti sottili di sequenza del DNA che possono essere usati per rilevare le varianti del gene colpevoli di una malattia. Durante questi ultimi anni, i ricercatori hanno usato gli SNPs per identificare varie regioni cromosomiche che contengono i geni in grado di influenzare il rischio di ottenere DMLE. Nel marzo 2006, tre gruppi indipendenti condotti da Josephine Hoh di Yale; Albert Edwards di Dallas e Lindsay Farrer di Boston, hanno segnalato quello che avevano scoperto su un gene del cromosoma 1 che aumenta notevolmente il rischio di ottenere DMLE. Il gene codifica per una proteina denominata Fattore H del Complemento che mantiene sotto stretto controllo il sistema del complemento in modo che non attacchi le cellule sane. I ricercatori hanno scoperto che persone con una variante particolare del gene per il fattore H erano più predisposte a sviluppare DMLE. La variante ad alto rischio potrebbe spiegare fino al 50% dei casi, presumibilmente perché il prodotto della proteina del gene alterato è meno efficace nell’inibizione della via del complemento. I geni correlati alla degenerazione maculare senile confermano la nozione che l’infiammazione abbia un ruolo centrale nel danneggiare la zona centrale della retina in questo disordine della visione. Hageman et al. hanno segnalato il collegamento tra un gene e l’attivazione della cascata del complemento. Questo gene, individuato sul cromosoma 6, produce una proteina denominata fattore B addetta all’attivazione del complemento.
L’infezione può essere determinante nello sviluppo di DMLE. Alcuni studi condotti su occhi di pazienti con DMLE essudativa, caratterizzata dallo sviluppo di vasi sanguigni nella macula, mostravano pazienti con pregressa infezione da Chlamydia. Una connessione di causa fra l’infezione data dalla Chlamydia e la DMLE deve essere tuttavia confermata.
Pericak-Vance et al. inoltre propongono l’interazione tra il gene LOC387715 e il tabagismo come uno dei fattori di rischio ambientali più importanti per la DMLE. I ricercatori hanno trovato che il rischio risultante dall’associazione ¶di fumare e trasportare la variante del gene era più alto della somma del rischio dei singoli due fattori nel promuovere lo sviluppo di DMLE.
Il fattore H e i geni di B e LOC387715 non sono gli unici fattori genetici che probabilmente influenzano il rischio di sviluppare DMLE. Per esempio, in uno studio pubblicato a gennaio, Pericak-Vance et al. si sono basati sull’analisi di otto geni che sono stati ritenuti coinvolti nello sviluppo di DMLE. La loro analisi ha anche compreso il VEGF. Il prodotto del gene VEGF stimola lo sviluppo dei vasi sanguigni, e ciò ha suggerito che esso possa partecipare alla formazione della DMLE essudativa, che è la forma più severa. Ma i geni del complemento ed il LOC387715 sono certamente i responsabili principali del rischio di DMLE. Dovendo basarsi su tali ricerche si può allora considerare seriamente l’uso di misure preventive come evitare di fumare, diminuire l’ingestione di cibi grassi ed aumentare l’assunzione di antiossidanti e di carotenoidi.
IDENTIFICAZIONE DEL FATTORE H
Hageman et al. sostengono che una variazione nel gene per il fattore H (HF1/CFH) accresce drammaticamente la probabilità di sviluppare la DMLE, così come la glomerulonefrite membrano-proliferativa di tipo II (MPGN II). HF1 codifica per una proteina coinvolta nella prima linea di difesa immunologica dell’organismo (il sistema innato) rivolta contro le infezioni batteriche e altri microbi. Di questo gene esisterebbero sia varianti protettive che a rischio.
In studi precedenti Hageman, Mullins, Anderson e Johnson implicavano la cascata del complemento, facente parte del sistema immunologico innato, nella formazione delle drusen. Le drusen contengono residui di EPR, processi cellulari dendritici e una varietà di molecole immuno-associate come immunoglobuline, antigeni di classe II, diverse componenti del complemento, attivatori e regolatori. Uno di questi regolatori, il fattore H, è una componente chiave della via alternativa di attivazione del complemento. Tutte insieme queste osservazioni hanno condotto i ricercatori a concludere che la DMLE, così come altre patologie senili, come il morbo di Alzheimer e l’aterosclerosi, possa veder coinvolta una grossa componente infiammatoria.
Gli Autori hanno notato che la MPGN II, se si tralascia il suo esordio precoce, ha manifestazioni oculari indistinguibili dalla DMLE. Una mutazione puntiforme nel gene HF1 (I1166R) causa MPGN II nei maiali e nei topi con una grave deficienza di fattore H, che sviluppano una gromeluronefrite severa. Inoltre gli individui affetti in due grandi famiglie colpite da MGPN III mostravano linkage al cromosoma 1q31-32, un locus vicino alla regione 1q25-31 che era stata precedentemente associata alla DMLE. Tutte queste osservazioni hanno portato i ricercatori a considerare il fattore H come il candidato principale per entrambe la DMLE e l’MPGN II. La variante genetica a rischio per lo sviluppo di DMLE più frequente è stata associata a quasi la metà dei 900 soggetti affetti in studio, mentre compariva nel 29% dei 400 controlli (P=10-3). La forza di questa associazione è di gran lunga superiore in confronto ad anomalie genetiche precedentemente attribuibili alla DMLE (ABCA4, FBNL5, FBNL6, geni APOE).
Nella prima linea di difesa contro i microrganismi e altre particelle estranee si trova il sistema del complemento, deputato a riconoscere, attaccare e uccidere i microrganismi invasivi creando dei fori nelle loro membrane. In alcuni casi, tuttavia, l’attivazione sostenuta del complemento può condurre all’infiammazione cronica, aggravare il danno tissutale locale e contribuire in modo significativo alla progressione della patologia, come succede nel morbo di Alzheimer e nell’aterosclerosi. Per prevenire tale danno alcune proteine, tra cui il fattore H, il principale inibitore solubile della via alternativa di attivazione del complemento, tengono il sistema sotto stretto controllo. Poiché la maggior parte delle variazioni identificate occupano importanti siti funzionali della proteina HF1, i ricercatori hanno suggerito la possibilità che queste varianti a rischio possano alterare il comportamento della proteina HF1 e ostacolare il suo ruolo nel regolare la via del complemento nella risposta immune.
Si potrebbe quindi affermare che individui con DMLE e MPGN II condividono un difetto funzionale nella proteina fattore H che colpisce la funzionalità del sistema del complemento. Variazioni di singoli nucleotidi potrebbero per esempio cambiare il legame di HF1 al frammento C3b del complemento o alla proteina C reattiva, all’acido sialico, o all’eparina. In modo simile queste variazioni potrebbero alterare la interazioni tra HF1 e i microbi, rendendo forse i tessuti, come l’EPR, più suscettibili alle infezioni. Questo concetto si basa sul fatto che i più importanti attivatori della via alternativa del complemento sono molecole che si ritrovano facilmente su molte superfici batteriche e virali. L’ipotesi finale consiste nel presupposto che una disfunzione del sistema del complemento possa risultare in un danno tissutale locale, soprattutto nei siti più vulnerabili come il glomerulo renale e la macula. Hageman et al. hanno anche dimostrato che lo strato elastico della membrana di Bruch è preferenzialmente più sottile nella regione maculare. Una rottura di questo fine strato e la conseguente infiammazione potrebbero spiegare la predilezione della macula alla formazione di lesioni, inclusa la formazione di membrane neovascolari coroideali.
L’aplotipo di HF1 a rischio si associa con una vasta gamma fenotipica di DMLE, eccetto forse l’atrofia geografica, osservata in un piccolo gruppo di pazienti; si presuppone quindi che non esista una relazione distinta tra il genotipo associato alla malattia e il fenotipo clinico.
Inoltre i risultati di questo studio sono congruenti con alcuni fattori di rischio epidemiologicamente associati alla DMLE. Il fumo ad esempio inibisce l’attività del fattore H e accresce il rischio di DMLE da 4 a 5 volte.
I dati di Hageman et al. concordano anche con le precedenti affermazioni sul ruolo del complemento nella DMLE. Sembra probabile che l’eredità di un aplotipo HF1 a rischio, in combinazione con un agente infettivo o altri attivatori atipici della via alternativa, come immunocomplessi, il peptide beta dell’amiloide, o il colesterolo, possano accrescere sostanzialmente la suscettibilità individuale alla DMLE e all’MPGN II.
È chiaro che le molecole coinvolte nell’attivazione del complemento e nella sua regolazione saranno i futuri target per lo sviluppo di test diagnostici precoci e trattamenti terapeutici per la DMLE e forse anche per altre patologie ad origine infiammatoria.
IL GENE HTRA1
Un locus sul cromosoma 10q26 è da tempo correlato al rischio di DMLE. Il polimorfismo di un singolo nucleotide, rs11200638, nella regione promotrice del gene HTRA1, è la variante genica più probabilmente coinvolta nello sviluppo della patologia, con un rischio attribuibile del 49,3%. Esperimenti immunoistochimici hanno svelato, tramite l’uso di anticorpi monoclonali, la presenza del prodotto di HTRA1 nelle drusen di occhi affetti da DMLE. Il polimorfismo del singolo nucleotide rs11200638 è situato 512 bp (paia di basi) a monte del sito di inizio della trascrizione del gene HTRA1. Tale gene codifica per un membro della famiglia delle proteasi seriniche espresse nella retina e nell’EPR (nei topi). HTRA1 sembrerebbe regolare la degradazione dei proteoglicani della matrice extracellulare. Questa attività si pensa possa facilitare l’accesso degli enzimi degradanti la matrice, come le collagenasi e le matelloproteasi della matrice, ai loro substrati. La sovraespressione di HTRA1 può alterare l’integrità della membrana di Bruch, favorendo l’invasione dei capillari coroideali attraverso la matrice extracellulare, come si verifica nella DMLE essudativa. Inoltre HTRA1 lega e inibisce il fattore di crescita trasformante ? (TGF-?) un importante regolatore della deposizione della matrice extracellulare e dell’angiogenesi. DeWan et al. riportano la stessa associazione tra tale variazione nucleotidica di HTRA1 e la DMLE essudativa nella popolazione cinese.
TIMP-3 nella membrana di Bruch: cambiamenti durante l’invecchiamento e nella DMLE
Le metalloproteasi della matrice (MMPs) e i rispettivi inibitori tissutali (TIMPs) giocano un importante ruolo nel ricambio della membrana extracellulare (ECM). Le MMPs costituiscono una famiglia di enzimi secernenti, attualmente con più di 20 membri, che sono coinvolti nel degradare componenti dell’ECM nel corso del normale ricambio e rinnovamento. Le MMPs, inoltre, sono implicate nelle fasi iniziali della neovascolarizzazione, in cui si pensa possano essere richieste, con altre proteasi, per la degradazione dei componenti della membrana capillare, come un requisito preliminare per la formazione di nuovi vasi. I TIMPs, che sono rappresentati da quattro prodotti distinti del gene, si pensa che sopprimano la degradazione eccessiva della ECM e possano svolgere un ruolo funzionale importante nella limitazione della neovascolarizzazione. Timp-3 una volta secreto, si lega a componenti della ECM, mentre le altre TIMPs no.
Un ruolo del Timp-3 nella membrana di Bruch può essere quello di potente inibitore locale dell’attività del MMP, regolare il turnover della membrana di Bruch, così come limitare la neovascolarizzazione coroidale.
La distrofia del fondo di Sorsby è paragonabile ad un inizio precoce, ereditario, della degenerazione maculare, caratterizzato dall’ispessimento della membrana di Bruch e dalla neovascolarizzazione submaculare. Mutazioni nel gene Timp-3 sono state trovate in famiglie con la distrofia del fundus di Sorsby. Studi di Immunoistochimica in un occhio donato da un paziente con Sorsby hanno mostrato un vasto accumulo di Timp-3 nella membrana di Bruch ispessita. Queste osservazioni hanno condotto a valutare la presenza del Timp-3 e la sua distribuzione nella membrana ispessita di Bruch degli occhi con DMLE. Anche se nessuna mutazione nella regione di codificazione o negli elementi regolatori del gene Timp-3 è stata scoperta in pazienti con DMLE fin qui osservati, Timp-3 in eccesso all’interno della ECM potrebbe essere causa di ispessimento della membrana di Bruch ed impedire il normale rimodellamento della membrana. A causa dell’ importanza della permeabilità della membrana di Bruch nel traffico dei metaboliti fra coroide e EPR, è importante capire il ruolo del Timp-3 nell’invecchiamento normale e nella DMLE.
Lo studio condotto da Motohiro Kamei e Joe G. Hollyfield ha analizzato 36 occhi normali enucleati post mortem (tra 14-96 anni) e 15 occhi con DMLE (tra i 74-98 anni) ed è stata valutato il cambiamento nella distribuzione del Timp-3 in relazione all’età. Sono stati confrontati anche i livelli di TIMP-3 negli occhi di pazienti con DMLE e nei controlli sani della stessa età.
Dallo studio si è visto che il Timp-3 immunoreattivo era presente nella membrana di Bruch in ogni campione di tessuto proveniente da donatori normali ed era distribuito nell’intero spessore della membrana. Anche se l’immunoreattività era uniforme in ogni campione dei donatori, in generale, gli occhi dei donatori più giovani hanno mostrato minore intensità di immunoreattività rispetto agli occhi dei donatori più anziani. Il Timp-3 immunoreattivo non era evidente nella retina neurosensoriale, in coroide, o nella sclera.
Distribuzione e presenza del TIMP-3 negli occhi con DMLE
Ciascuno occhio con DMLE utilizzato conteneva soft drusen. Ogni volta che sono state osservate drusen morbide, sia isolate che confluenti, ciascuna era intensamente immunoreattiva con l’anticorpo anti- Timp-3 . Anche le drusen dure, quando presenti, erano fortemente immunoreattive.
Nella membrana di Bruch sotto le distese zone di atrofia dell’EPR, l’immunoreattività Timp-3 non era evidente o poco rilevabile. La neovascolarizzazione coroidale era presente in 8 dei 15 occhi di donatori con DMLE. Nelle zone dove l’EPR aveva proliferato intorno ad una membrana neovascolare coroidale o a una cicatrice fibroblastica, il Timp-3 immunoreattivo circondava l’EPR iperplastico. L’immunoreattività era bassa e mostrava una distribuzione irregolare, probabilmente a causa di una perdita della funzione e della polarità della proliferazione dell’EPR.
Nelle regioni di transizione fra le zone di atrofia dell’EPR e un EPR normale, vi erano diminuzioni di immunoreattività del Timp-3, ma esso era presente nella drusen morbide e sotto lo strato elastico centrale della membrana di Bruch.
La quantità di Timp-3 presente nella membrana di Bruch sotto la macula nella retina umana normale sembra essere dipendente dall’età.
Sezioni provenienti dalla fovea di donatori dalla seconda alla terza decade di vita erano debolmente immunoreattive con l’anticorpo anti-TIMP-3. Inoltre, la distribuzione di Timp-3 è cambiata nella nona e decima decade, con l’estensione dell’ immunoreattività dalla membrana di Bruch alla coriocapillare.
L’analisi quantitativa ha mostrato che il contenuto e la funzione del Timp-3 nella macula aumentava con l’età con un significativo coefficiente di correlazione (r = 0.66 e 0.67, rispettivamente). Timp-3 quindi è una proteina correlata all’invecchiamento. L’analisi quantitativa ha indicato che i livelli Timp-3 erano significativamente elevati nella macula degli occhi con DMLE rispetto ad occhi normali.
Gli occhi con DMLE, tuttavia, hanno mostrato una distribuzione non uniforme, con una virtuale assenza di immunoreattività di Timp-3 nelle zone di atrofia dell’EPR e una abbondante immunoreattività nelle zone al di fuori delle aree atrofiche dove l’EPR era presente. Questo indica che la distribuzione del Timp-3 nella DMLE in occhi con atrofia dell’EPR non è uniforme.
Vari fattori ereditari o non ereditari quali alterazioni proteiche, sforzi ossidativi, o disordini degli enzimi idrolitici possono contribuire, con l’invecchiamento, ad accelerare l’accumulo di questi residui. Poiché Timp-3 inibisce largamente le MMPs, drusen con un eccesso di Timp-3 possono ritardare il rinnovamento della membrana di Bruch. Ciò può provocare ispessimento della membrana di Bruch, riduzione della permeabilità della membrana di Bruch al traffico di metaboliti e di sostanze nutrienti fra la coroide ed l’EPR, e conseguente atrofia dell’EPR e dei fotorecettori.
Nelle zone dove era stata osservata neovascolarizzatione coroidale, l’EPR era assente e nessuna immunoreattività per Timp-3 era evidente nella membrana di Bruch sottostante.
Concludiamo che il contenuto di Timp-3 nella membrana di Bruch in macula aumenta durante il normale invecchiamento e che il contenuto di Timp-3 è elevato oltre i livelli normali nella regione maculare degli occhi con DMLE. Ciò suggerisce che Timp-3 possa essere una delle molecole chiave nell’ispessimento della membrana di Bruch nell’invecchiamento normale ed nella DMLE.
Indicatori sistemici di infiammazione, disfunzione endoteliale e DMLE
È stato ipotizzato che l’infiammazione abbia un ruolo nella patogenesi della DMLE. Hageman et al. hanno indicato che le drusen contengono proteine connesse con processi immuno-mediati ed infiammazione. Cellule infiammatorie croniche sono state osservate sulla superficie esterna della membrana di Bruch in occhi con degenerazione maculare senile di tipo essudativo. Queste cellule possono causare lesioni aterogeniche e microvascolari da rilascio diretto di ossidanti, composti tossici dell’ossigeno ed enzimi proteolitici che possono anche danneggiare la membrana di Bruch. Tuttavia, i dati degli studi epidemiologici fin qui osservati, non hanno indicato un rapporto costante fra infiammazione sistemica o presenza di markers infiammatori e DMLE.
La disfunzione endoteliale, derivata dall’infiammazione, dall’ipertensione, dal tabagismo e da altri fattori, è stata considerata un presupposto nella patogenesi della DMLE. L’intento dello studio di Klein et al. è stato di analizzare se i markers di infiammazione sistemica e di disfunzione endoteliale fossero associati alla DMLE. Le procedure seguite in questo studio hanno incluso: il peso, l’altezza e la pressione sistemica, un questionario standardizzato, che ha incluso domande specifiche per quanto riguarda una storia di enfisema, artrite, tabagismo, consumo di alcol ed uso di vitamine.Non sono state trovate prove dell’associazione di questi indicatori di infiammazione sistemica con la DMLE, in accordo con i dati dello studio cardiovascolare (CHS). Questa mancanza di associazione tra gli indicatori di infiammazione sistemica studiati (hsCRP sierica, SAA, Tnf-? ed IL-6) e DMLE concorda con l’inefficacia dimostratasi nell’uso di farmaci antinfiammatori per via sistemica. Prove cliniche hanno invece suggerito i possibili effetti benefici dei corticosteroidi somministrati per via intravitreale. I corticosteroidi intravitreali riducono l’incidenza di membrane neovascolari laser-indotte nei primati, attraverso la riduzione dell’attività delle cellule infiammatorie e del loro numero nella coroide. Altri potenziali meccanismi includono la riduzione dell’espressione del VEGF e la down-regulation della molecola di adesione 1 intracellulare (ICAM-1) espressa sulle cellule dell’EPR e sull’endotelio dei vasi, che regola l’adesione dei leucociti e la diapedesi durante l’infiammazione.
CONCLUSIONI
Cinque concetti riassuntivi rilevanti nella patologia della DMLE
Primo: le alterazioni legate all’età con l’aggiunta di eventi patologici determinano DMLE.
Secondo: lo stress ossidativo negli anni e nella DMLE causa danno all’EPR e alla coriocapillare.
Terzo: il danno dell’EPR e della coriocapillare nella DMLE deriva da un’infiammazione cronica della coroide e della membrana di Bruch.
Quarto: il danno all’EPR, alla coriocapillare e l’infiammazione possono condurre ad anomalie della matrice extracellulare (ECM). Questa ECM anormale causa un’alterazione della diffusione degli elementi nutritivi dall’EPR alla neuroretina, che può provocare ulteriori danni retinici e all’EPR.
Quinto: la anomalie alla matrice extracellulare (ECM) portano ad una alterazione della funzione del complesso EPR-coriocapillare che evolve verso l’atrofia retinica, l’atrofia dell’EPR e la crescita di nuovi vasi (CNV).
In questa sequenza di eventi l’ambiente e la genetica possono modificare il decorso della patologia.
Dott.ssa Laura Bertazzi
Dott. Cristiano Luiselli
Dott. Pierfilippo Sabella
Cattedra di Clinica Oculistica
Dipartimento di Scienze Cliniche “Luigi Sacco” – Milano
Unità Operativa Di Oculistica Ospedale “Luigi Sacco” – Milano
BIBLIOGRAFIA
1. Klein R et al. Prevalence of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology 99: 933-943, 1992.
2. Kahn HA et al. The Framingham Eye Study Outline and major prevalence findings. Am J Epidemiol 106: 17-32, 1977.
3. Seddon JM, George S, Rosner B, Rifai N. Progression of age-related macular degeneration: prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch Ophthalmol. 2005 Jun;123(6):774-82.
4. Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006 Mar-Apr;51(2):137-52.
5. Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004 Apr;122(4):598-614.
6. Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001 Nov;20(6):705-32
7. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration: the involvement of immunocompetent cells. Graefes Arch Clin Exp Ophthalmol. 1985;223(2):69-76.
8. Forrester JV. Macrophages eyed in macular degeneration. Nat Med 2003 Nov;9(11):1350-1
9. Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003 Nov;9(11):1390-7.
10. Wiggs JL. Complement factor H and macular degeneration: the genome yields an important clue. Arch Ophtalmol. 2006 Apr;124(4):577-8.
11. Despriet DD, Klaver CC, Witteman JC, Bergen AA, Kardys I, de Maat MP, Boekhoorn SS, Vingerling JR, Hofman A, Oostra BA, Uitterlinden AG, Stijnen T, van Duijn CM, de Jong PT. Complement factor H polymorphism, complement activators, and risk of age-related macular degeneration. JAMA. 2006 Jul 19;296(3):301-9.
12. Haines JL, Schnetz-Boutaud N, Schmidt S, Scott WK, Agarwal A, Postel EA, Olson L, Kenealy SJ, Hauser M, Gilbert JR, Pericak-Vance MA. Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest Ophthalmol Vis Sci. 2006 Jan;47(1):329-35.
13. Emil Wirostko, William J. Wirostko, Barbara M. Wirostko. Age-related macular degeneration is an inflammatory disease possibly treatable with minocyclineActa Ophthalmol Scand. 2004 Apr;82(2):243-4.
14. Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006 Nov 10;314(5801):992-3.
15. Kamei M, Hollyfield JG. TIMP-3 in Bruch’s membrane: changes during aging and in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1999 Sep;40(10):2367-75.
16. Bailey TA, Alexander RA, Dubovy SR, Luthert PJ, Chong NH. Measurement of TIMP-3 expression and Bruch’s membrane thickness in human macula. Exp Eye Res. 2001 Dec;73(6):851-8.
17. Strunnikova N, Hilmer S, Flippin J, Robinson M, Hoffman E, Csaky KG. Differences in gene expression profiles in dermal fibroblasts from control and patients with age-related macular degeneration elicited by oxidative injury. Free Radic Biol Med. 2005 Sep 15;39(6):781-96
18. Nakata K, Crabb JW, Hollyfield JG. Crystallin distribution in Bruch’s membrane-choroid complex from AMD and age-matched donor eyes. Exp Eye Res. 2005 Jun;80(6):821-6.
19. Bok D. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc Natl Acad Sci U S A. 2005 May 17;102(20):7053-4.
20. Penfold PL, Killingsworth MC, Sarks SH. Senile macular degeneration. The involvement of giant cells in atrophy of the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1986 Mar;27(3):364-71.
21. Penfold P, Killingsworth M, Sarks S. An ultrastructural study of the role of leucocytes and fibroblasts in the breakdown of Bruch’s membrane. Aust J Ophthalmol. 1984 Feb;12(1):23-31.
22. Vine AK, Stader J, Branham K, Musch DC, Swaroop A.Biomarkers of cardiovascular disease as risk factors for age-related macular degeneration. Ophthalmology. 2005 Dec;112(12):2076-80.
Dr. Carmelo Chines
Direttore responsabile